









Introduction
Honey has been recognized as a wholesome food which may enhance culinary life style and provide therapeutic merits, e.g., recovery from physical debilitation, because it is a sugary food with various pleasing fragrances from various floral plants and provides nutrients and functional substances such as antioxidants including phenolic compounds and flavonoids and minerals as well as simple saccharides. However, since some of beekeepers and honey wholesalers have committed to illegal adulteration of their honey products for extra profit and sale boost, the quality and authenticity of honey have attracted attention from consumer and beekeeping industry as well as relevant government authority. Honey frauds include mislabeling of floral origin and honeybee species, adulteration of honey with inverted sugar and various syrups (White et al., 1986; Wu et al., 2017), and production of honey by feeding bees with commercial industrial sugar (Guler et al., 2014).
Apis cerena, called the Asiatic honeybee is a species of honeybee which has been traditionally used for beekeeping in Korea, but Korean native honey (KNH) from A. cerena is produced much less than European honey (EH) from the European honeybee, A. mellifera. Due to imbalance of supply and demand, the price of KNH is five to seven times more expensive than EH. Thus, some bee-keepers and honey sale companies may mislabel EH as KNH and distribute the fake KNH.
The different electrophoretic and immunological characteristics of the major proteins in KNH and EH were used to detect the fraud of KNH (Lee et al., 1998; Won et al., 2008; Won et al., 2009). The molecular weights of the major proteins in KNH and EH were 56 and 59 kDa, respectively, which enabled sodium dodecyl sulfate–polyacrylamide gel electrophoresis to become a detection method. However, the small difference of 3 kDa in molecular size did not allow precise identification of polypeptide bands in SDS-PAGE (Lee et al., 1998; Won et al., 2008). Won et al. (2009) reported that the immunological method could also be applied to detect the fraud. Their preparations of polyclonal antibodies against the major proteins from KNH and EH did not cross-react with EH and KNH, respectively.
Recently, DNA-based molecular techniques based on polymerase chain reaction (PCR) were recognized as quick, accurate, specific methods to identify animal species of food products and thus to detect adulteration/substitution of food (Bottero et al., 2011; Kumar et al., 2015; Salihah et al., 2016).
Mitochondrial genes have been targeted preferentially for molecular diagnostics of animal species including honey bee (Dalmasso et al., 2004; Herbert et al., 2003; Kek et al., 2017; Processer and Hebert, 2017; Xue et al., 2017). The animal mitochondrial DNA exhibits rapid evolution relative to nuclear gene and has high copy number in cells, so it can be used as a sensitive tool to differentiate among animal species. PCR seems to be the best of DNA-based methods in species identification and furthermore, multiplex PCR allows discrimination of several species at the same time (Bottero et al., 2011). Though real-time PCR which can shorten analysis time without additional electrophoresis step (Salihah et al., 2016), it requires high-priced real-time PCR instrument which need to be maintained periodically for proper function of the machine. Therefore, it would be desirable to develop a simple, rapid, sensitive method, which does not require the expensive real-time PCR instrument and electrophoresis procedure.
Immunochromatographic assay (IC), also known as lateral flow test, is a simple device to detect the presence of a target substance for medical diagnostics without need for the costly equipment. The IC can be an alternative, fast, cost-effective technique for detection of PCR-amplified DNA with digoxigenin and biotin at the 5’-ends. The PCR-amplified DNA which is applied to a nitrocellulose filter in IC diffuses through the nitrocellulose filter to pick up gold-streptavidin conjugate and then reaches to the location of immobilized anti-digoxigenin antibody. The resulting complex of gold-streptavidin conjugate, PCR-amplified DNA, and anti-digoxigenin antibody shows up as a black-colored band at the location. For multiplex PCR, fluorophores, such as fluorescein isothiocynate, carboxytetramethylrhodamine, and so forth, can be used to label the additional primers.
The objective of this study is to develop a simple, rapid system of duplex PCR and IC to discriminate between KNH and EH.
Materials and Methods
Honey products were obtained from local beekeepers and purchased at internet shopping sites in Korea. Accuprep genomic DNA purification kit, Gold multiplex PCR premix, and primer oligonucleotides (Bioneer, Korea), Wizard genomic DNA purification kit (Promega, USA), Chelex 100 (Bio-Rad, USA), NuSieve 3:1 agarose (Lonza, USA), monoclonal anti-TAMRA antibody, rabbit antidigoxigenin antibody (Abscam, USA), streptavidine (NE Biolab, USA), and gold nanoparticles (Rapigen, Korea) were used in the study. The kit for immunochromatography assay (IC) was assembled and provided by Rapigen in Korea. All other chemicals were reagent grade.
For the Chelex resin method, five gram of honey was dissolved in 45 mL of distilled water. The honey solution was centrifuged at 3,500 g for 30 min. After supernatant was discarded, the pellets were washed with 1 mL of TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0). To the washed pellets, twenty mililiter of Chelex resin suspension (5 g Chelex 100, 50 mL Tween 20, 100 mL TE buffer), 8 mL of proteinase K (10 mg proteinase K, 1 mL of TE buffer), and 180 mL of TE buffer were added. The mixture was reacted at 56°C for 30 min, heated at 100°C for 8 min, and then centrifuged at 10,000 g for 10 min. The supernatant was saved and stored at −20°C.
For the Wizard genomic DNA purification kit method, to the washed pellet prepared same as in the Chelex resin method, 600 mL of nuclei lysis solution of the kit was added. Two hundred mililiter of protein precipitation solution was added to the pellet suspension. The mixture was vortexed for 20 s, cooled in iced water for 5 min, and then centrifuged at 10,000 g for 4 min. Six hundred mililiter of the resultant supernatant was taken and added to 600 mL of isopropyl alcohol in a new tube. The mixture was centrifuged at 10,000 g for 5 min and the supernatant was discarded. The pellet was washed with 70% ethanol and then dissolved in 200 mL DNA rehydration solution. The solution was stored at −20°C.
For the Accuprep genomic DNA extraction kit method, to the washed pellet, 200 mL of PBS and 20 mL of proteinase K in the kit were added. The subsequent procedures followed the kit manual provided by Bioneer. The DNA extract which were bound to binding column of the kit was finally eluted by using 200 mL of elution buffer of the kit and stored frozen at −20°C.
Primers specific for Apis mellifera and Apis cerana were designed based on its mitochondrial cytochrome C oxidase subunit I gene sequences from Genebank KJ396191and DQ020245 (Table 1). The forward primers, M5-F and C6-F, which were labeled on 5'-end with digoxigenin and carboxytetramethylrhodamine (TAMRA), respectively and the reverse primers, M5-R and C5-R, which were labeled on 5'-end with biotin were used for IC. To the reaction mixture tube of Gold multiplex PCR premix, 0.5 mL of genomic DNA, 1 mL of primers (10 mM), and sterilized distilled water were added to make up to 20 mL. The PCR reaction was performed in the DNA Engine Gradient Cycler (Bio-Rad, USA). The PCR procedure was 94°C for 5 min followed by 35 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 30 s and finally 72°C for 5 min. The PCR products were analyzed by using agarose gel electrophoresis in 3% NuSieve 3:1 agarose gel in TAE buffer with ethidium bromide staining. The PCR products amplified with the primers labeled with digoxigenin, TAMRA, and biotin were analyzed by using IC.
Gold nanoparticle was prepared by reduction of gold chloride with carbonate. Gold-streptavidine (SA) conjugate was prepared as follows; One mililiter of Gold nano-particle solution (~40 nm in diameter) was adjusted at pH 6.0 and 60 mL of SA (0.5 mg/mL) were added slowly and stirred for 1 h. For blocking gold-SA conjugate, A hundred mililiter of 5% bovine serum albumin (BSA) were added and mixed briefly for 30 min. After centrifugation at 10,000 g for 1 h, the supernatant was removed and suspended in 100 mL of solution containing 5% BSA, 0.1% polyethyleneglycol, and 50 mM sodium phosphate, pH 7.2.
In order to prepare test strip for immunochromatographic assay, the gold-SA conjugate was jetted onto a conjugate pad using BioDot dispenser (Bio-Rad, USA) and then dried at 37°C for 3 h. The monoclonal anti-TAMRA antibody, rabbit anti-digoxigenin antibody and rabbit anti-SA antibody were immobilized on the nitrocellulose filter as two test lines and a control line, respectively and dried at 30°C for 3.5 h. The test strip was placed in a plastic kit which had a hole for sample pad and a slit for viewing results.
The mixture containing 10 mL of PCR product and 90 mL of PBST (PBS with 0.05% Tween 20) was added onto the sample pad of test strip. After 5 min, the bands of gold nanoparticle complex were observed visually and photographed.
Results and Discussion
Genomic DNA was extracted from 5 g of a Korean native honey (KNH) and a European honey (EH) by using Chelex resin method, Wizard DNA purification kit, and Accuprep DNA extraction kit to look for an efficient DNA extraction method for duplex PCR. The DNA preparations from the three procedures were used for duplex PCR where the EH-specific primers for A. mellifera, M2-F and M2-R, and the KNH-specific primers for A. cerana, C6-F and C5-R, were added to reaction solutions. Amplified DNA products were detected by agarose gel electrophoresis with ethidium bromide staining (Fig. 1). The agarose electrophoresis in Fig. 1 showed that duplex PCR with the genomic extracts from KNH and EH produced DNA of 178 bp and 206 bp, respectively, as described in Table 1. The intensity of amplified DNA obtained from the DNA extract by using the Chelex resin method appeared the strongest of all. These results suggested that the Chelex resin method is simple, rapid, and efficient. The Chelex resin method was used for following experiments in this study.
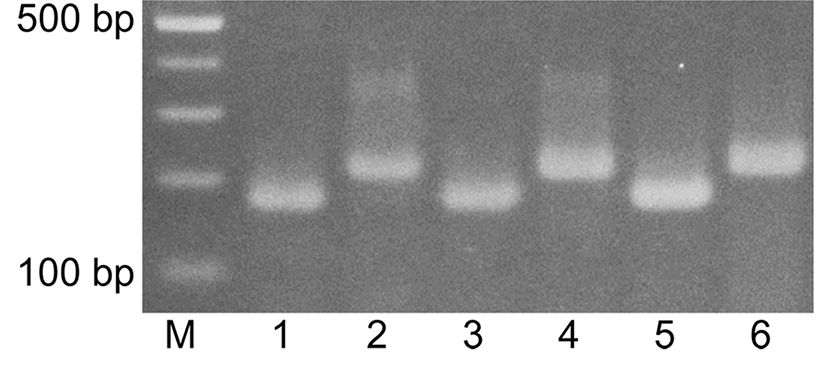
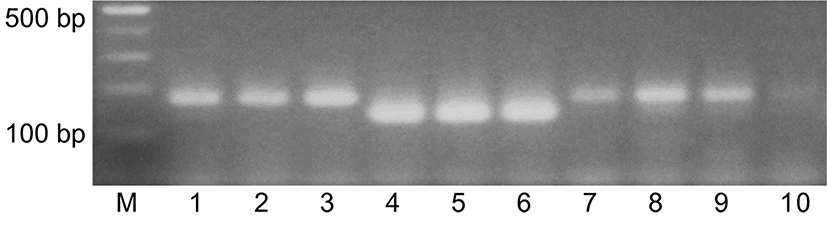
A set of mixtures of KNH and EH with different ratios as described in Fig. 2 was prepared and genomic DNA was extracted from the mixtures. The genomic DNA extracts from the mixed honeys were used for duplex PCR where the EH-specific primers, M2-F and M2-R, and the KNH-specific primers, C6-F and C5-R, were added to the reaction solutions. Agarose gel electrophoresis in Fig. 2 showed that DNA of 206 bp was detected from the mixtures of down to 5% EH but not from the mixture of 1% EH. An additional set of EH-specific primers, M5-F and M5-R, was designed to improve sensitivity in duplex PCR. Agarose gel electrophoresis in Fig. 3 showed that DNA of 133 bp was detected from the mixtures of down to 1% EH, when M5-F and M5-R was used in the duplex PCR. These results indicated that the primers, M5-F and M5-R were more sensitive than M2-F and M2-R in detecting EH from the mixture in the duplex PCR.

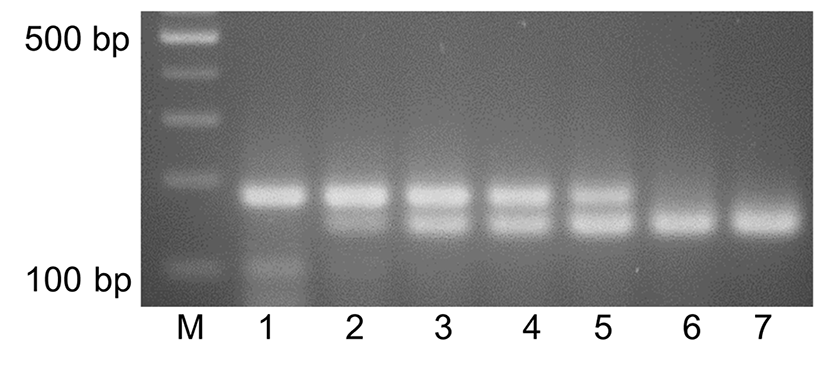
Seven KNH products from local beekeepers and three KNH products from internet shopping sites were used for identification of the honeybee species of the KNH products by using duplex PCR and agarose gel electrophoresis. The genomic DNA extracts from the honey products were used for duplex PCR where the EH-specific primers, M5-F and M5-R, and the KNH-specific primers, C6-F and C5-R, were added to the reaction solutions. Agarose gel electrophoresis in Fig. 4 showed that DNA of 177 bp was amplified from seven KNH products from local beekeepers (lanes 1-3 and 7-10) and DNA of 133 bp was amplified from the three KNH (lanes 4-6) from internet shopping sites, which suggested that the apparent KNH products from internet shopping sites were actually EH products. Two of KNH products at lanes 7 and 10 showed weak DNA bands of 178 bp, which seemed to suggest that the quality of the genomic DNA extract was not appropriate for the duplex PCR. Similar results have been shown in duplex PCR where EH-specific primers, M2-F and M2-R instead of M5-F and M5-R, was used (results not shown).
These results indicated that the duplex PCR with subsequent agarose gel electrophoresis could identify fake KNH-labeled honey products which were actually EH and also detect EH which is mixed with KNH at low ratio. However, since some of KNH products in lanes 7 and 10 of Fig. 4 showed weak DNA bands, more improvement in preparation method of genomic DNA extract from a couple of KNH products is required in future application.
Sensitivity of immunochromatographic assay (IC) in detecting of DNA which was amplified in duplex PCR from the genomic DNA extracts from the mixture of KNH and EH was examined. For the IC, the forward primers, M5-F and C6-F were labeled on 5`-end with digoxigenin and TAMRA, respectively. The reverse primers, M5-R and C5-R were labeled on 5`-end with biotin. The labeled primers were used in the duplex PCR. The IC in Fig. 5 showed that EH-specific bands appeared for the mixture containing 5% EH. The KNH-specific band was weaker than the EH-specific band and did not show up for 10% KNH in the IC. All kits showed bands at the location of streptavidine (SA), which indicated that gold-SA conjugate left over after reaction with PCR-amplified DNA showed immunological reaction at the location of immobilized anti-SA antibody and evidenced that gold-SA conjugate diffused from the conjugate pad to the location of the immobilized anti-SA antibody in the IC kit as expected.

Four KNH products from local beekeepers and three KNH products from internet shopping sites were used for identification of the honeybee species of the KNH products by using duplex PCR and IC (Fig. 6). The duplex PCR was performed with the EH-specific primers, M5-F and M5-R, and the KNH-specific primers, C6-F and C5-R. The IC at kits 1-3 and kits 4-6 in Fig. 6 showed KNH-specific bands and EH-specific bands, respectively, which indicated that the KNH products from internet shopping site were not KNH but EH. No band showed up for the KNH product at kit 7 of Fig. 6. The KNH product which showed no band at the kit 7 of Fig. 6 was the same honey that showed a relatively weak DNA band at the lane 7 of Fig. 4.

These results showed that the IC could replace agarose gel electrophoresis for detecting duplex PCR products. Even though the IC was less sensitive than agarose gel electrophoresis in detecting PCR-amplified DNA, it was still an appropriate technique to detect EH with relatively good sensitivity. Considering that more than 2-3 h are required to run a set of agarose gel electrophoresis, the IC which can show the results at least within 5 min may be a simple, rapid method to detect PCR-amplified DNA in the discriminating test of KNH and EH.
However, overall low reaction of KNH-specific band in the IC might result in no band for certain KNH products, which appeared at the kit 7 of Fig. 6. When no band showed up in the IC, accurate determination of the bee species of the honey product could not be made. Additional studies are required to improve repeatability of the genomic DNA extraction method from various KNH products and to enhance sensitivity of the PCR and IC in detecting KNH.
Specificity, sensitivity, and convenience of the PCR-based assay to discriminate between Korean native honey and European honey was determined by comparing among DNA preparation methods, primers, and detection methods in this study. The species-specific primer sequences based on cytochrome C oxidase gene of the two honey bee species were designed by placing at least a mismatch at the 3’ terminus of the primer, as described by Kwok et al. (1995). Additional introduction of two to three mismatches to the primer was, if possible, sought to achieve greater level of discrimination and to enhance repeatability among assays. Then specificity and sensitivity of the designed primers were determined by performing PCR assays by using different primer sets in this study. The PCR assays were designed to detect European honey sensitively, which may prevent commercial distribution of fake Korean native honey, restore consumer’s confidence, and promote consumption of genuine Korean native honey. The PCR assay was also simpler and more sensitive than the nucleotide sequencing techniques which were used to determine honey entomological origin by Kek et al. (2017) and Processer and Hebert (2017), but could not still be used for on-site inspection of honey. Further developments of simple procedures and portable instruments are required for the easy, quick examination of honey bee species in future.