










Introduction
Yogurt is one of the most popular fermented food products, and its consumption is thought to be beneficial to human health (Shiby and Mishra, 2013). Yogurt is traditionally produced through fermentation by lactic acid bacteria (LAB), including Lactobacillus delbrueckii subsp. bulgaricus and Streptococcus thermophilus. bulgaricus and Streptococcus thermophilus are used. Yogurt also often contains probiotics, such as Lactobacillus acidophilus, Lacticaseibacillus casei and Bifidobacteria spp. (Heller, 2001; Kulp and Rettger, 1924; Lourens-Hattingh and Viljoen, 2001). Dairy companies use different combinations of these LAB starters or probiotics to give their yogurt products specific flavors and textures. However, the LAB often continue their lactic acid fermentation during the product’s shelf life, and this phenomenon is known as “post-acidification”. The phenomenon causes several adverse effects on yogurt products, such as too much acidic taste, decrease of viable LAB count and higher syneresis, which ultimately reduces the shelf life of the product (Abu-Jdayil and Mohameed, 2002; Donkor et al., 2006; Lubbers et al., 2004; Vinderola et al., 2000). Post-acidification increases the size of casein particles, hydrophobic and electrostatic interactions between proteins, and consequently causes restructuring of the protein network (re-curding), increasing viscosity of the products (Deshwal et al., 2021). The point is that lactic acid fermentation continues even with cold chain and the situation is much worse in tropical regions or developing countries with poor refrigeration systems. Therefore, this phenomenon needs to be controlled to maintain good quality of yogurt during transportation and storage.
Several methods have been suggested for the control of post-acidification, including adding preservative such as benzoic or sorbic acids, nisin, vanillin, bacteriocin, changing the ratio of Lactobacillus spp. and Streptococcus spp., modifying milk composition, applying pulse electric field, and using weak post-acidification bacteria or genetically modified bacteria as starter (Chanos et al., 2020; Dave and Shah, 1997; Han et al., 2012; Zhang et al., 2011). However, these methods have not provided satisfiable solution for post-acidification. Even though genetically modified bacteria have obvious potential, it still has trouble to be used in food due to consumer’s negative perception (Bawa and Anilakumar, 2013; Maghari and Ardekani, 2011). Lately, Vieira et al. (2021) showed positive correlation between accumulation of bioactive amines in cow’s fermented milk and post-acidification which further emphasizes the importance of controlling post-acidification. Consequently, the post-acidification is still a problem to be solved.
Lactic acid fermentation can be simply described as a metabolic process that produces cellular energy and the lactic acid, from glucose utilization. Glucose is converted into two molecules of pyruvate through glycolysis, and subsequently into lactate by lactate dehydrogenase (LDH) in the presence of the cofactor nicotinamide adenine dinucleotide + hydrogen (NADH). For the LAB, this reaction is important in the concept of regeneration of NAD+, which is needed in early fermentation stage as the oxidizing agent. Therefore, LDH is considered as a key enzyme of lactic acid fermentation, and control of this enzyme could be one of the solutions to prevent post-acidification.
Different species of LAB produce D- and L-lactic acid with different ratio (Kandler and Weiss, 1986; Schleifer, 1986). In the case of L. casei, L-LDHs are important for lactic acid fermentation because L. casei produces L-lactic acid predominantly (Vijayakumar et al., 2008). The L-LDHs of L. casei need fructose 1,6-diphosphate (FDP) for their proper activities (Hensel et al., 1983), and these allosteric L-LDHs are reported to possess much higher activities than the non-allosteric ones (Jiang et al., 2014).
In this sense, the objectives of this study were to identify the L-LDHs produced by L. casei HY2782; to clone and overexpress these enzymes in Escherichia coli BL21 (DE3) strain; and characterize enzymatic properties of the L-LDHs.
Materials and Methods
E. coli strain DH5α and BL21 (DE3) were purchased from BioFACT (Daejeon, Korea) and Invitrogen (Carlsbad, CA, USA), respectively. The cloning vector pGEM-T Easy was from Promega (Madison, WI, USA) and the expression vector pET22b (+) was from Merck KGaA (Darmstadt, Germany). Whole-genome sequencing of L. casei HY2782 and annotation were conducted by the ChunLab Whole Genome Analysis Service (ChunLab, Seoul, Korea). Deoxyribonucleic acid (DNA) extraction and purification kit from Qiagen (Hilden, Germany), enzymes from Takara Bio (Shiga, Japan) and T4 DNA ligase from Roche (Basel, Switzerland) were used for DNA cloning. The nikel-nitrilotriacetic acid (Ni-NTA) agarose column was purchased from Qiagen. All the medium for microbial growth were purchased from Difco (Franklin Lakes, NJ, USA) and all chemicals were from Sigma-Aldrich (St. Louis, MO, USA).
L. casei HY2782 was supplied by HY (Seoul, Korea). L. casei HY2782 was inoculated into MRS broth at 37°C anaerobically. E. coli DH5α and BL21 (DE3) were inoculated into Luria-Bertani (LB) broth with shaking or LB agar (1.5% agar, w/v) at 37°C. These strains were preserved in 10% (w/v) skim milk supplemented with 25% (v/v) glycerol at –80°C. pGEM-T Easy and pET22b (+) were used to construct expression vector (Table 1).
Four L-LDH genes (Supplementary Fig. S1) amplified from genomic DNA (Supplementary Fig. S2) of L. casei HY2782 were inserted into the pGEM-T Easy vector, a linearized vector with a single 3′-terminal thymidine at both ends (TA cloning; Holton and Graham, 1991). The constructed pJE vectors (Table 1, Fig. 1) were transformed into E. coli DH5α. Ampicillin (Amp, 100 μg/mL) and 5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal, 40 μg/mL) were supplemented in the LB agar for blue/white screening for E. coli DH5α harboring pJEs (Sambrook et al., 1989a). The pJEs were extracted from the recombinant strains (white colonies) and inserted into pET22b (+) vector through NdeI/XhoI digestion and ligation. Each constructed pET22b-ldhL (Table 1, Fig. 1) was transformed into E. coli BL21 (DE3) respectively.
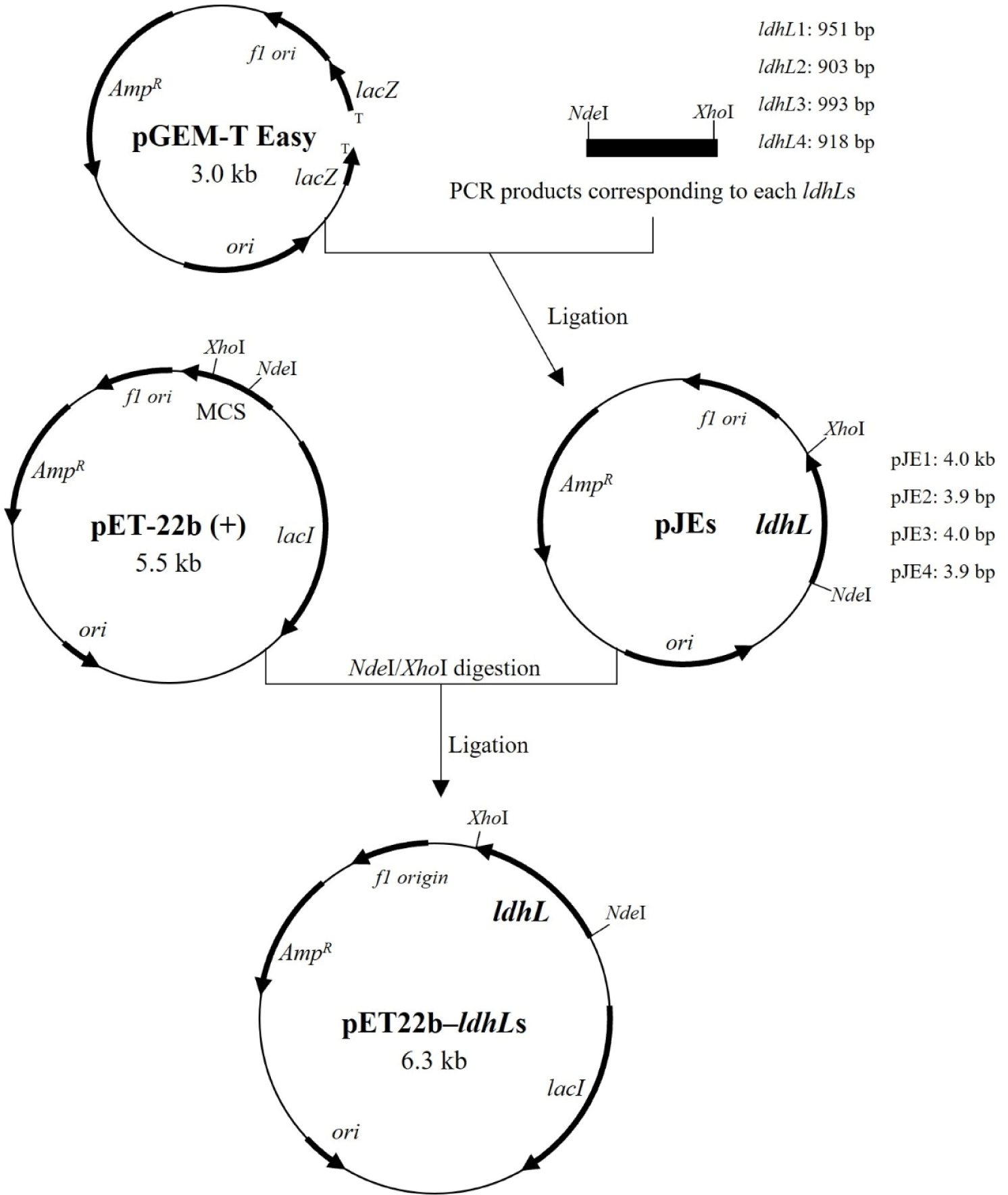
The recombinant E. coli BL21 strains were grown in baffled flask containing LB broth supplemented with Amp (100 μg/mL) at 37°C and 150 rpm for 3 h, the optical density at 600 nm (OD600) reached about 0.6. To induce the expression of ldhL genes, isopropyl-β-D-thiogalacto-pyranoside (IPTG) was added to a final concentration of 1 mM. After incubation under each condition (Table 2) for each strain, the cells were collected by centrifugation and washed two times using 1× PBS buffer containing NaCl (8 g/L), KCl (0.2 g/L), Na2HPO4 (1.44 g/L), and KH2PO4 (0.24 g/L). Cell pellets were resuspended in same buffer and disrupted by sonication (pulse on, 2 s; pulse off, 10 s; 58% amplitude; VCX 500, Sonics, Newtown, CT, USA) on ice for 6 min, four cycles. Disrupted cells were centrifuged and the supernatants were filtered using syringe with 0.45 μm filter.
Each cell lysate were loaded on Ni-NTA agarose column, and loaded samples were washed two times using wash buffer (Table 3). Then the L-LDHs were eluted using elution buffers containing different concentration of imidazole (Table 3). The fractions containing eluted proteins were identified using 12% (w/v) SDS-PAGE. To identify approximate molecular weight of the proteins, Precision Plus ProteinTM Dual Color Standards (Bio-Rad, Hercules, CA, USA) were loaded together. Electrophoresis was conducted at 100 V for 1 h and 40 min. The gels were stained in staining buffer (Table 3) with shaking for 1 h. Then the stained gels were destained in destaining buffer (Table 3) with shaking for 4 h and further destained in distilled water overnight (Sambrook et al., 1989b). The fractions indicated single band on SDS-PAGE were pooled. The purified proteins were concentrated and buffer-changed by centrifugation using Amicon Ultra (30 kDa, Merck Millipore, Burlington, MA, USA), two times with 10 mM Tris-HCl buffer (pH 8.0) containing 1 mM ethylenediamine-tetraacetic acid (TE buffer, Bioneer, Daejeon, Korea).
The L-LDH activity for conversion of pyruvate to lactate was analyzed by measuring the reduction rate of NADH at 340 nm using UV spectrophotometer (Cary 300 UV VIS, Agilent Technologies, USA). The enzyme reaction mixture contained citrate-citric acid buffer (50 mM, pH 4.0), 10 mM sodium pyruvate, 0.2 mM NADH, and 8 mM FDP in a total volume of 3 mL. The reactions were conducted at 43°C and initiated by adding each enzyme (Jiang et al., 2014).
One unit was defined as the amount of enzyme required to catalyze the oxidation of 1 μmol of NADH per min. Beer-Lambert law was used to convert absorbance to concentration. Specific activity was expressed as units per mg of protein (Powers et al., 2007).
The optimal pH for L-LDH3 activity was determined in following buffers for each pH condition: 50 mM citrate-citric acid buffer (pH 3.0–6.5; 0.5 pH unit intervals), 50 mM Tris-HCl buffer (pH 7.0–8.0; 0.5 pH unit intervals), and 50 mM carbonate-bicarbonate buffer (pH 9.0–11.0; 1.0 pH unit intervals). The enzyme reactions were conducted as described previously at 43°C with 8 mM FDP. The effect of pH on the enzyme stability was investigated using the enzyme pre-incubated at 37°C for 1 h with different pH buffer (pH 3.0–11.0; 1.0 pH unit intervals), same buffers with specific pH that were used for optimal pH assay, and the reaction was conducted at 43°C in citrate-citric acid buffer (50 mM, pH 4.0) with 8 mM FDP.
The optimal temperature for L-LDH3 activity was determined by conducting the enzyme reaction at following temperature with 8 mM FDP: 4°C, 10°C, 25°C, 30°C, 37°C, 43°C, 50°C, 55°C, 60°C, and 65°C. The effect of temperature on the L-LDH3 stability was examined using the enzyme pre-incubated at different temperatures (25°C, 37°C, 43°C, 50°C, 55°C, 60°C, and 65°C) for 10 min in citrate-citric acid buffer (50 mM, pH 6.0).
The relative activity was determined by the highest specific activity as 100%. Each experiment was conducted in triplicate.
To investigate the effect of metal ions on L-LDH3 activity, MgSO4, CaCl2, MnSO4, ZnSO4, and CuSO4 were added to the reaction mixture at final concentration of 2 mM. The enzyme reaction was conducted at 43°C in citrate-citric acid buffer (50 mM, pH 4.0) with 10 mM sodium pyruvate, 0.2 mM NADH, 8 mM FDP, and 2 mM metal ions.
KH2PO4 was added to the reaction mixture at different final concentration (2, 4, 8, and 16 mM) to examine their inhibitory effects on L-LDH3 activity. The enzyme reaction was performed at 43°C in citrate-citric acid buffer (50 mM, pH 4.0) with 10 mM sodium pyruvate, 0.2 mM NADH, 8 mM FDP and different concentration of KH2PO4.
The relative activity was determined by the control (without metal ions or KH2PO4) as 100%. All experiments were conducted in triplicate.
All experiments were conducted in triplicate, and the results were expressed as means and standard deviations. Statistical significance (p<0.05) was determined using one-way analysis of variance (ANOVA; IBM SPSS Statistics 30, IBM, Armonk, NY, USA) along with Duncan’s multiple range test.
Results and Discussion
The recombinant strains of E. coli BL21 harboring pET22b-ldhLs were induced to express of each L-LDH after IPTG induction at each condition (Table 2). Then the expressed proteins were purified using Ni-NTA agarose column. The expression and purification of the recombinant protein were identified by SDS-PAGE analysis (Fig. 2). The molecular weights of L-LDHs was approximately 30–37 kDa on SDS-PAGE.
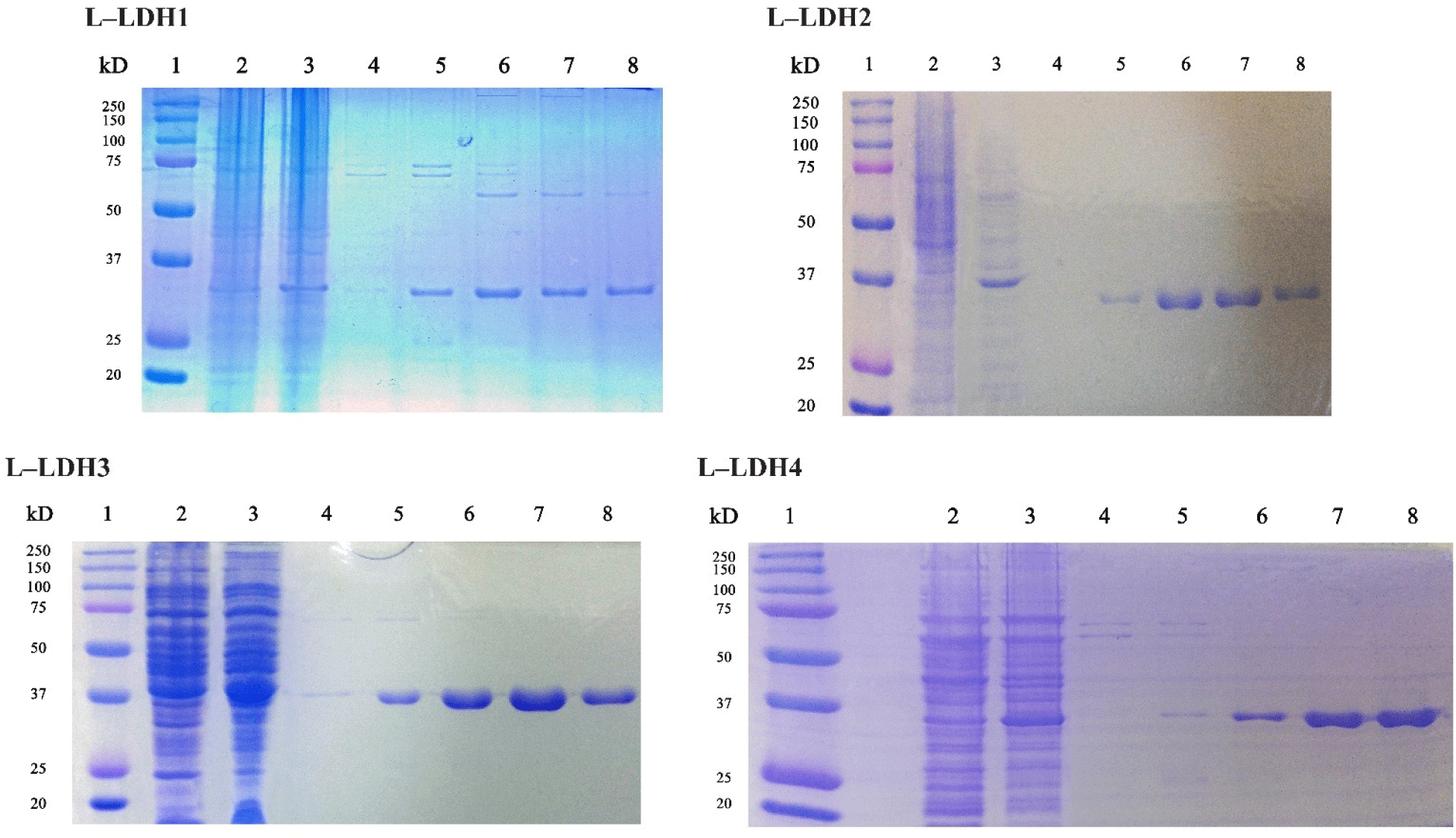
About the L-LDH1, the fractions eluted by elution buffers containing 150, 200, and 250 mM imidazole (Fig. 2) were pooled and concentrated using Amicon Ultra. First, the enzyme reaction was conducted at 25°C in Tris-HCl buffer (50 mM, pH 7.0) with 10 mM sodium pyruvate, 0.2 mM NADH and 5 mM FDP. Absorbance at 340 nm was recorded every 2 min, however, no reduction was observed until 30 min (Fig. 3).
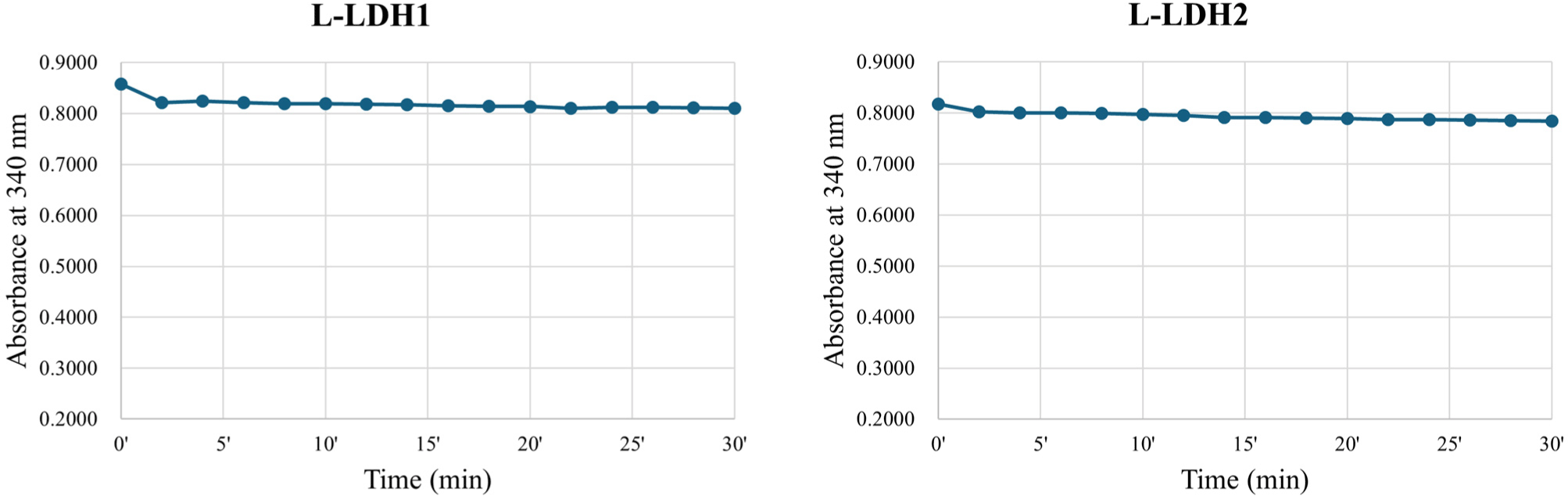
The L-LDH2 fractions eluted by elution buffers containing 150, 200 mM imidazole were pooled and concentrated (Fig. 2). The enzyme activity was examined at same condition for L-LDH1. The L-LDH2 also exhibited no activity (Fig. 3).
The fractions of purified L-LDH3 eluted by elution buffers containing 150, 200 mM imidazole were pooled and concentrated by centrifugation with Amicon Ultra (Fig. 2). The enzyme reaction mixture was prepared as described above. The reduction of absorbance at 340 nm was recorded every 2 s (Fig. 4). The specific activity of purified L-LDH3 was 3,990.42 U/mg.
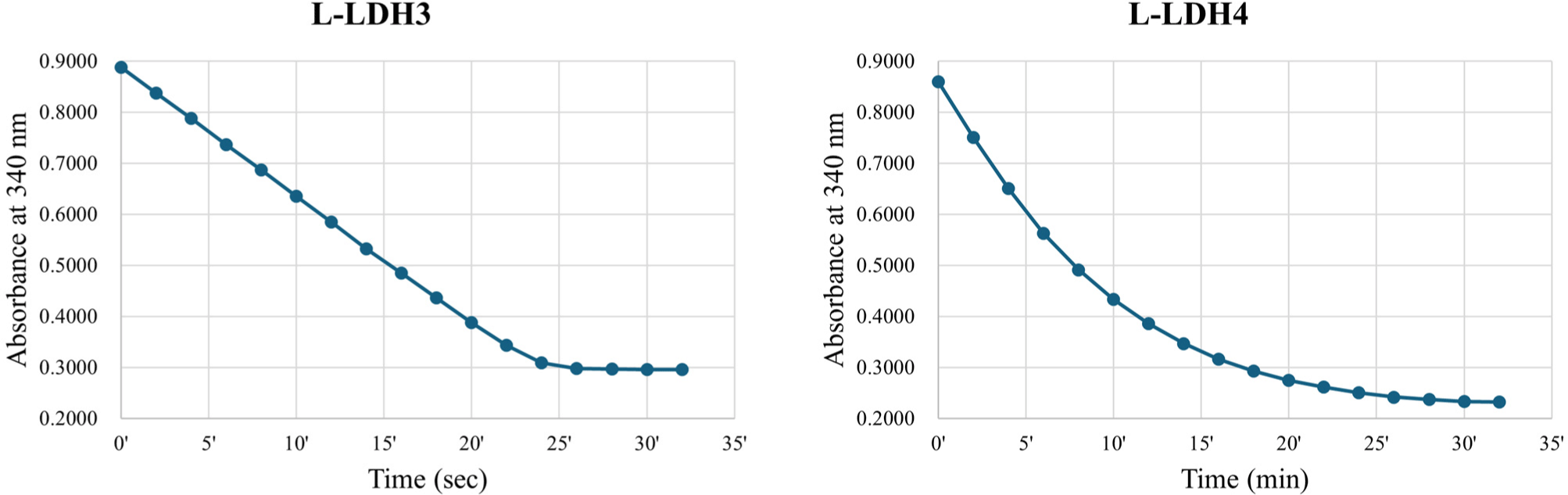
Purified fractions of L-LDH4 eluted by elution buffers containing 200, 250 mM imidazole were pooled and concentrated (Fig. 2). The enzyme reaction was performed as described before. Absorbance at 340 nm of reaction mixture was recorded every 2 min (Fig. 4). Purified L-LDH4 exhibited 165.82 U/mg of specific activity.
To summarize, the L-LDH3 showed the highest enzyme activity among four L-LDHs of L. casei HY2782 at 25°C with 5 mM FDP. Although L-LDH4 also indicated activity at same condition, the specific activity was almost 4% of that of L-LDH3. Other two L-LDHs, L-LDH1 and L-LDH2, exhibited no activity under this condition. Consequently, L-LDH3 seems to be a major L-LDH of L. casei HY2782.
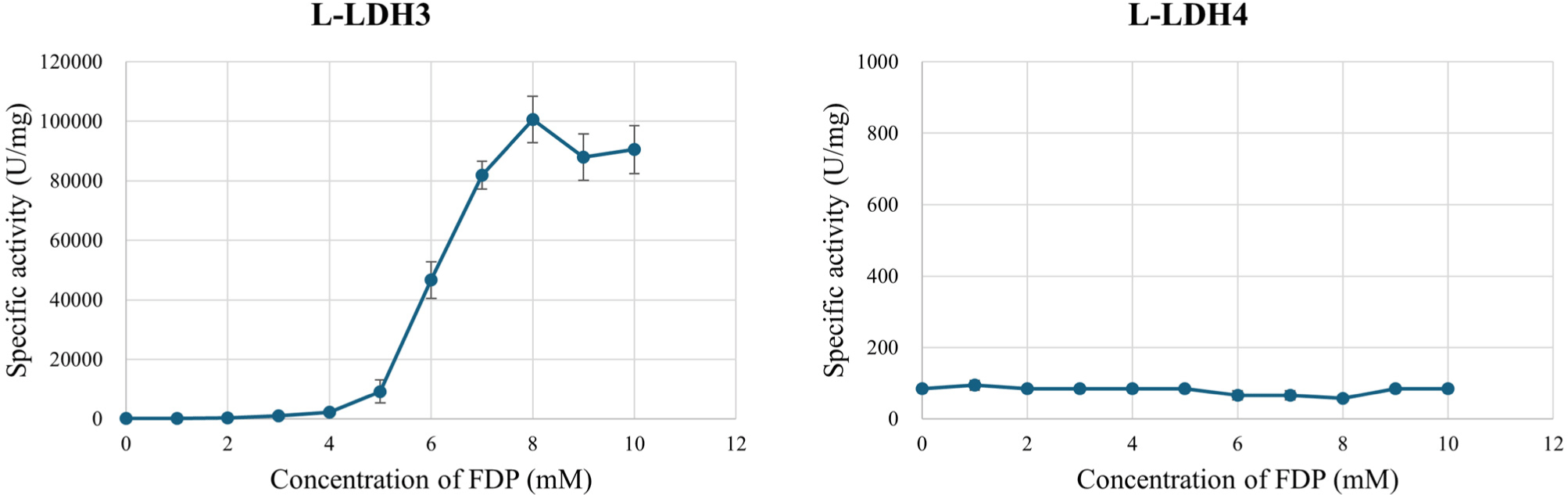
To verify whether the L-LDHs are FDP-dependent allosteric enzyme or not, effect of FDP on L-LDH3 and L-LDH4 activities was examined (Fig. 5). Different concentrations of FDP (0–10 mM at 1 mM unit intervals) was added to enzyme reaction mixture comprising Tris-HCl buffer (50 mM, pH 7.0) with 10 mM sodium pyruvate, 0.2 mM NADH. Without FDP in enzyme reaction mixture, L-LDH3 exhibited specific activity by 112.04 U/mg that was similar to the L-LDH4. The specific activity increased as concentration of FDP increased to 8 mM. With 8 mM FDP, L-LDH3 indicated the highest specific activity by 30,736.99 U/mg. Increasing the concentration of FDP over 8 mM did not increase the activity further. The results indicate that the L-LDH3 is an allosteric enzyme that needs FDP for their proper activities which are similar to previous studies (Arai et al., 2002; Arai et al., 2010; Hensel et al., 1983). In contrast, enzyme activity of L-LDH4 was unaffected by the concentration of FDP. The specific activity maintained a constant level from 56.58 to 94.30 U/mg. The result that L-LDH3 showed higher activities compare to non-allosteric L-LDH4 is consistent with Jiang et al. (2014) report, which allosteric L-LDH possess much higher activities than the non-allosteric ones.
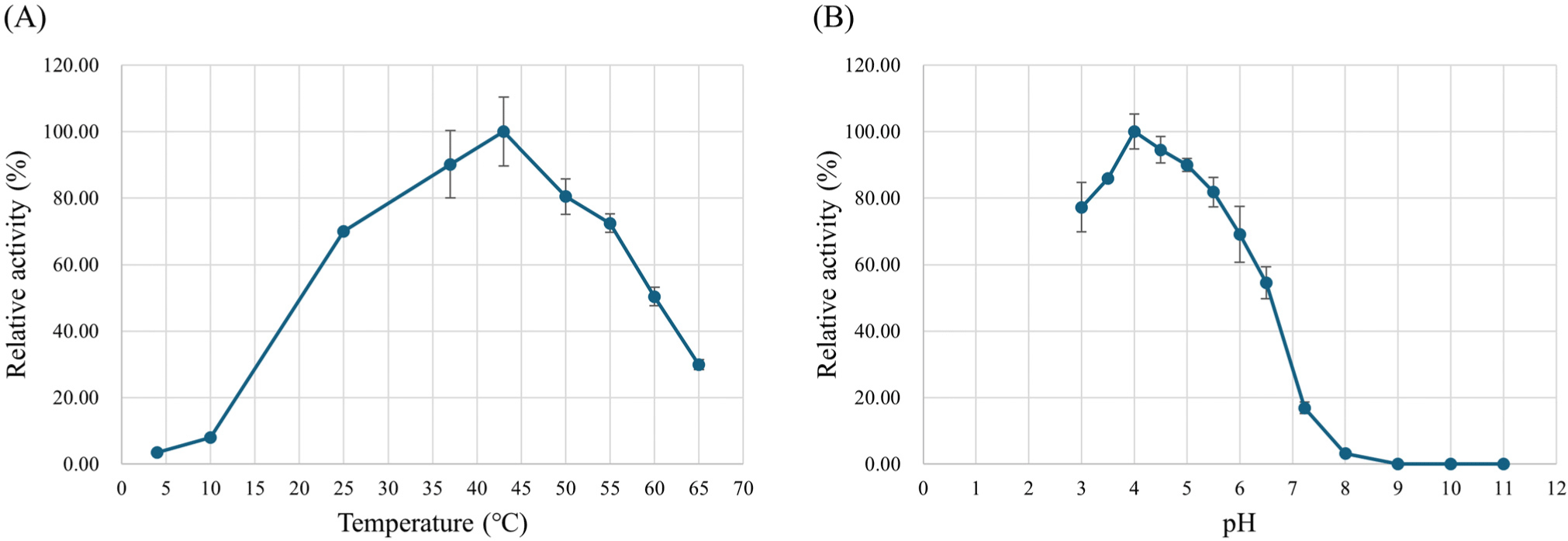
Various pH values (pH 3.0–11.0) were tested to determine the optimal pH for L-LDH3 activity. The maximal activity of L-LDH3 was observed to be 302,343.16 U/mg at pH 4.0. The activity increased drastically from pH 4.5 to 4.0 and decreased at pH 3.5 (Fig. 6A). Although the optimal pH for L-LDH activity of L. casei HY2782 was much lower than that from previous studies (pH 5.0–5.5), it is similar in that it shows higher activity in acidic conditions (Holland and Pritchard, 1975; Mayr et al., 1980). It was described that L-LDH of L. casei indicates higher sensitivity to FDP and exhibits marked activity even without FDP under acidic conditions (Arai et al., 2001; Arai et al., 2002; Garvie, 1980; Hensel et al., 1977; Hensel et al., 1983; Holland and Pritchard, 1975; Mayr et al., 1980). Arai et al. (2010) explained the high pH-dependence in the allosteric effects in relation to the structure of L. caseiL-LDH, which is the unique intersubunit salt bridges in the active state. Therefore, the higher activity at below the pH 5.0 could be due to the higher FDP sensitivity. The optimal temperature was determined by comparing the activity at various temperatures (4°C–65°C). The L-LDH3 exhibited maximal activity at 43°C (Fig. 6B).
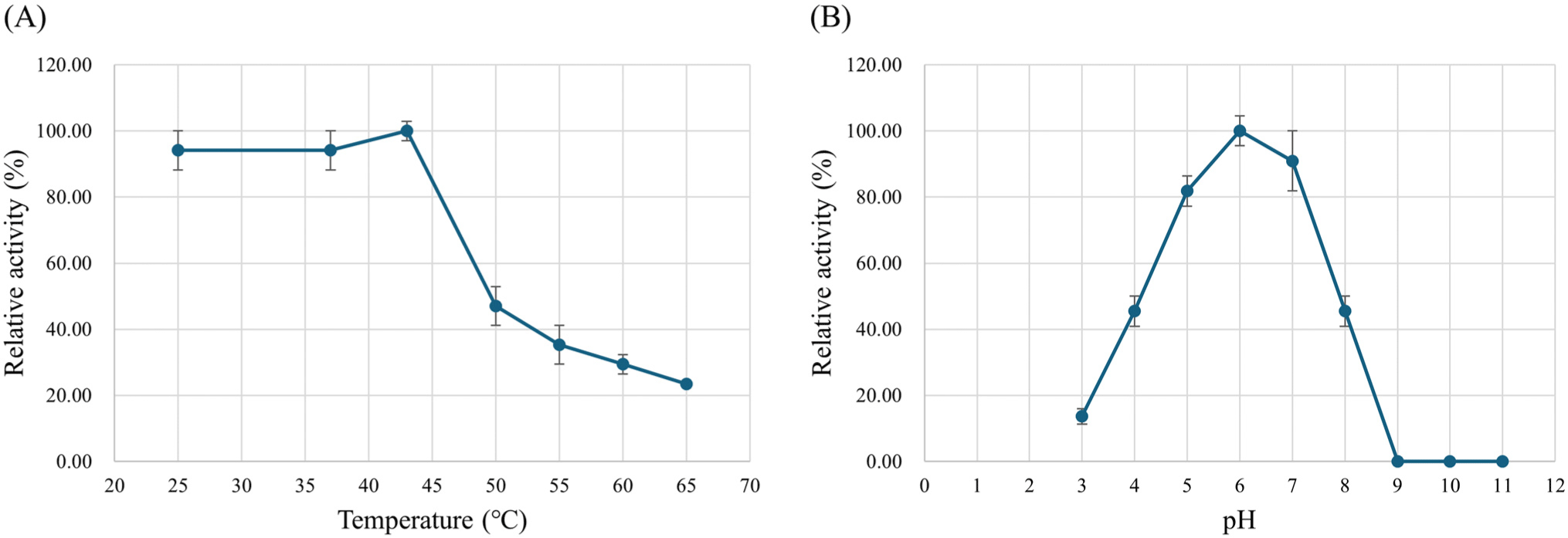
The enzyme stability was investigated at different pH (pH 3.0–11.0) and temperature (25°C–65°C) values. L-LDH3 retained more than 80% of the enzyme activity after incubation at pH 5.0–7.0 and 45% was retained after incubation at pH 4.0 and 8.0 (Fig. 7A). Effect of temperature on the enzyme stability was assessed by pre-incubation at pH 6.0 and different temperature for 10 min. The enzyme activity rapidly decreased after incubation at 50°C and over (Fig. 7B).
The enzyme activity with various metal salts was examined by adding each metal ion to the reaction mixture at final concentration of 2 mM. The reaction was conducted at 43°C, pH 4.0 with 8 mM FDP. In various bacterial L-LDHs, manganese (Mn2+) exhibited a positive effect on enzyme activity (Crow and Pritchard, 1977; Hensel et al., 1977). According to the Holland and Pritchard (1975), Co2+, Cd2+, Cu2+, and Ni2+ ions activated the L-LDH of L. rhamnosus as effective as Mn2+ at pH 6.3, Fe2+ and Zn2+ were less effective but also activated the L-LDH, Mg2+ exhibited no activation effect. In this study, Mg2+, Ca2+, and Mn2+ indicated positive effect on L-LDH3 of L. casei HY2782, especially Ca2+ was more effective than Mn2+, and Zn2+, Cu2+ ions inhibited the activity of L-LDH3 (Fig. 8A). The results are consistent with several previous studies that the activation of L. caseiL-LDH markedly improved in the presence of certain divalent metal ions such as Mn2+, Mg2+ and Ca2+ at acidic condition (pH 5.0–5.5; Furukawa et al., 2014; Mayr et al., 1980). Under pH 6.5, on the other hand, Zn2+, Cu2+ showed positive effects like Mn2+, but not Mg2+ for L-LDH of L. casei (Holland and Pritchard, 1975). The differences between the studies may come from the difference of enzyme origin or assay conditions, especially pH. However, the information about the effect of metal ions at acidic condition could be more useful in view of control of L-LDH activity in fermented milk.
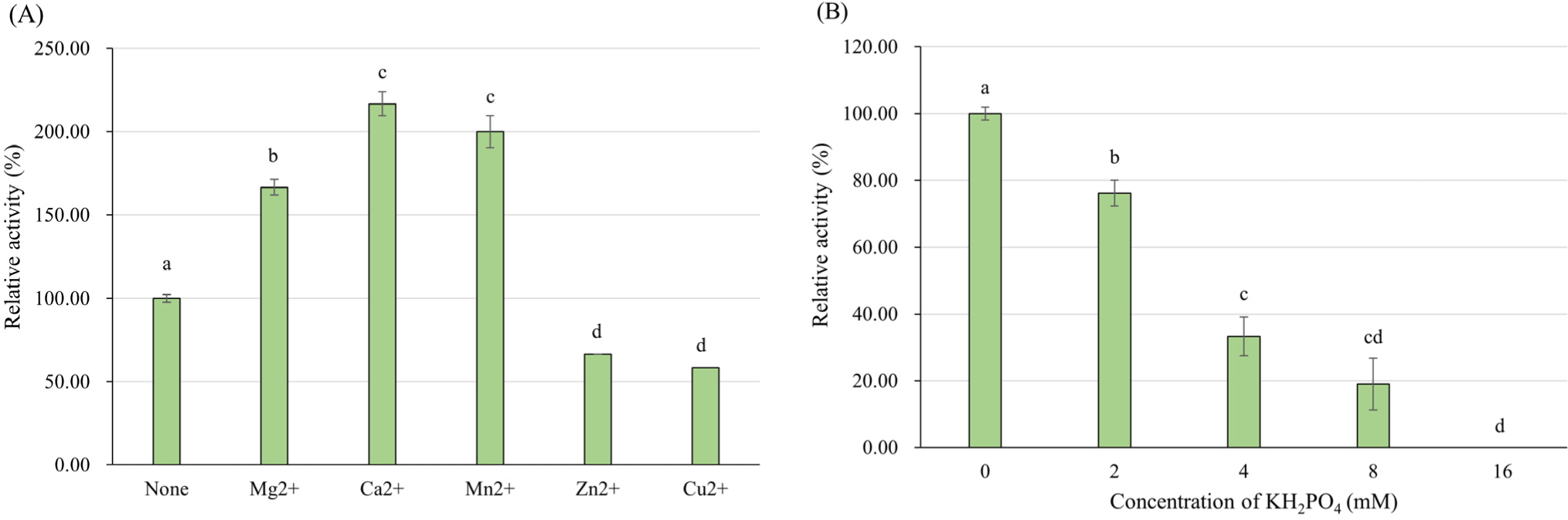
According to the results of amino acid sequence analysis (Supplementary Fig. S3) and effect of FDP concentration on enzyme activity, the L-LDH3 of L. casei HY2782 seems to be allosteric enzyme that activated by FDP. Several studies (Arai et al., 2002; Taguchi and Ohta, 1995) revealed that His-188 residue of allosteric L-LDH interacts directly with phosphate group of FDP. Therefore, phosphate is generally considered as competitive inhibitor for FDP-dependent L-LDHs (Anders et al., 1970; Crow and Pritchard, 1977; Garvie, 1978; Garvie, 1980; Garvie and Bramley, 1979a; Garvie and Bramley, 1979b; Neimark and Tung, 1973). However, the tendency of inhibition by phosphate is vary among L-LDHs of different bacteria. The sensitivity about FDP can affect the inhibitory effect of phosphate, and higher FDP sensitivity results in lower inhibition by FDP (Garvie and Bramley, 1979b). Some LDH could even be stimulated by phosphate (e.g. LDH of Streptococcus faecalis at neutral pH; Garvie, 1980). To verify whether phosphate inhibits L-LDH3 of L. casei HY2782, effect of KH2PO4 on L-LDH3 activity was investigated by adding different concentration of KH2PO4 in enzyme reaction mixture. Considering the concentration of FDP (8 mM), KH2PO4 concentrations for experiment were decided. The activity of L-LDH3 decreased to 19% of that of control (without KH2PO4) with 8 mM KH2PO4, same concentration as FDP. By adding 16 mM KH2PO4, the enzyme activity was completely inhibited (Fig. 8B).
Conclusion
Post-acidification is main obstacle to extending shelf-life of yogurt, which is still unsolved problem in yogurt industry. In the present study, we overexpressed and characterized L-LDHs of L. casei HY2782 to control the post-acidification caused by this strain. Four genes encoding L-LDH were identified from L. casei HY2782 by whole genome sequencing. These genes were successfully cloned and expressed in E. coli BL21 (DE3). Among the four L-LDHs, L-LDH3 exhibited the highest enzyme activity of 302,343.16 U/mg at 43°C and pH 4.0 in the presence of 8 mM fructose 1,6-bisphosphate. The enzyme activity was retained more than 80% after incubation at pH 5.0–7.0 and rapidly decreased after incubation at over 50°C. In acidic condition, Mg2+, Ca2+, and Mn2+ ions stimulated L-LDH3 activity, and Zn2+ and Cu2+ ions had inhibitory effects on L-LDH3. In addition, L-LDH3 was inhibited by KH2PO4. The results indicated that L-LDH3 may play an important role in lactic acid fermentation of L. casei HY2782 as major L-LDH of this strain. Therefore, control of L-LDH3 with potential inhibitors, Zn2+, Cu2+, and KH2PO4, could be one of the solutions to mitigate the post-acidification caused by L. casei HY2782. However, further study is needed to verify whether these inhibitors actually reduce the post-acidification. Nevertheless, this study provides some basic information to better understand the lactic acid fermentation of L. casei strains and could be new approach to control the post acidification in fermented dairy products such as yogurt.