










Introduction
Inflammation is an immune response that can be activated by various conditions and stimuli which involve the injury of tissue or infection (Medzhitov, 2008). Immune cells and inflammatory mediators are known to be involved in the inflammatory process. Nitric oxide (NO) is one of the inflammatory mediators that play a variety of roles in physiological and pathological immune responses (Chang et al., 2019). NO is synthesized by the nitric oxide synthase (NOS) family, which consisting neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Under normal conditions, nNOS and eNOS produce the required amount of NO that regulates cell proliferation and survival, while iNOS produces large amounts of NO to enhance immunity (Lundberg et al., 1997). However, the overproduction of NO has been associated with the progression of many diseases, including chronic inflammation, Alzheimer’s disease, diabetes mellitus, and cancer (Dali-Youcef et al., 2013; Sharma et al., 2007). Therefore, new compounds that inhibit the overproduction of NO are considered candidates for the production of novel anti-inflammatory agents.
Macrophages play a vital role in immune reactions, and the functions of macrophages that include phagocytosis, antigen presentation, and NO and inflammatory cytokine production (Lee et al., 2017b). Macrophages produce pro-inflammatory factors via several cell signaling pathways, including nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK) pathways when they are activated by lipopolysaccharides (LPS), which are one of the main sources of stimulation for macrophages (Ham et al., 2015; Li et al., 2017). The MAPK pathways have three different main families, which are Jun amino-terminal kinases (JNK) pathway, extracellular signal-regulated kinases (ERK) pathway, and p38 pathway (Ham et al., 2015). This has made the MAPK pathways the primary targets for the development of novel anti-inflammatory agents and has placed them at the center of this field of research (Seo et al., 2015).
Many peptides produced from various protein sources, such as soy (de Mejia and Dia, 2009), fish (Ahn et al., 2015) and lupine (Millán-Linares et al., 2014), are known to have anti-inflammatory properties. These peptides inhibit the production of pro-inflammatory cytokines and NO in immune cells after stimulation. de Mejia and Dia (2009) reported that peptides derived from soy proteins suppressed the NF-κB pathway and inhibited the production of inflammatory cytokines in LPS stimulated RAW 264.7 macrophages. Some egg proteins were also used to prepare anti-inflammatory peptides: Ovotransferrin peptides (IRW) inhibited the production of several cell adhesion molecules associated with the onset and progression of inflammation in human endothelial cells (Huang et al., 2010). Sun et al. (2016) reported that ovomucin peptides have anti-inflammatory activities by suppressing the NF-κB pathway. Ovalbumin (OVA) has important functional properties that make it useful for gelling, foaming, and emulsifying of food products. Many studies have shown that the hydrolysates or peptides from OVA possess various bioactivities, including antioxidants (Abeyrathne et al., 2014), antihypertensive (Matoba et al., 1999), and antimutagenic effects (Vis et al., 1998). However, few studies have assessed the anti-inflammatory activity of OVA hydrolysates in relation to their inhibitory effects on the production of NO in immune cells.
The objectives of present study were to determine the effect of OVA hydrolysates on the production of NO and the expression of iNOS mRNA, and elucidate the anti-inflammatory mechanism of OVA hydrolysates in LPS-stimulated RAW 264.7 cells.
Materials and Methods
OVA was isolated from egg white using the method of Abeyrathne et al. (2013). Briefly, egg white was diluted with an equal volume of distilled water, homogenized for 1 min using a hand-blender (Kitchen-Aid) at high speed (set at 9) and used. Lysozyme, ovomucin, and ovotransferrin were sequentially removed from the diluted egg white using the method developed by Abeyrathne et al. (2014). Lysozyme was removed first from the egg white solution: Amberlite FPC 3500 resin (5 g/100 mL diluted egg white) was added to the diluted egg white solution and stirred overnight in a 4°C walk-in cooler using an overhead stirrer set at 250 rpm (RW20 digital, IKA Works, Wilmington, NC, USA) to trap lysozyme. After filtering out the resins, the pH of the lysozyme-free egg white solution was adjusted to 4.75 using 3 N HCl to precipitate ovomucin. The precipitated ovomucin was removed by centrifugation at 3,500×g for 30 min at 4°C (Sorvall Evolution RC superspeed centrifuge, Thermo Scientific, Waltham, MA, USA), and then the lysozyme- and ovomucin-free egg white was added with ammonium sulfate (5.0%, wt/vol) and citric acid (2.5%, wt/vol) and held for 12 h at 4°C to precipitate ovotransferrin, which was removed by centrifugation at 3,500×g for 30 min. The resulting supernatant, which was mainly composed of OVA and ovomucoid, was added with 100% ethanol to the final concentration of 35%, and then centrifuged. After removing the residual ovomucoid using 4 volumes of 35% of ethanol, the resulting precipitant was washed with 4 volumes of distilled water twice, the pH adjusted to pH 12.0 to dissolve OVA, and then brought the pH down to 10.0 using 1N HCl. Finally, the dissolved OVA was heated at 70°C for 15 min to precipitate impurities, centrifuged, and then lyophilized using a freeze-dryer (Labconco, Kansas City, MO, USA). The purity and yield of the isolated OVA was 97% and 97.7%.
Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM), penicillin-streptomycin, and phosphate-buffered saline (PBS) were purchased from Hyclone Laboratories, Inc. (Logan, MI, USA). N-(1-naphthyl)-ethylenediamine, sulfanilamide, thiazolyl blue tetrazolium bromide (MTT), LPS, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). For PCR analysis, RNeasy Mini Kit for total RNA isolation was purchased from Qiagen (Milan, Italy), RevertAid™ First Strand cDNA Synthesis Kit was purchased from Thermo Fisher Scientific (Carlsbad, CA, USA), and SYBR reagent was purchased from PhileKorea (Daejeon, Korea). For Western blotting analysis, cell lysis buffer, inhibitor cocktail containing protease and phosphatase inhibitor, and a Protein Assay Kit were purchased from Thermo Fisher Scientific. Primary antibodies for total or phosphorous from MAPKs (p38, JNK, and ERK), iNOS, and β-actin were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). A secondary antibody (Goat anti-mouse IgG-HRP) and other reagents for western blotting were purchased from Bio-Rad (Hercules, CA, USA). All other chemicals used were of analytical reagent grade.
Because the functional activity of peptides varies depending on their amino acid sequence, length, and structural characteristics (Hartmann and Meisel, 2007), four different proteolytic enzymes were used in this study. OVA (10 mg/mL) were hydrolyzed using one of the following proteases: Trypsin (from bovine pancreas, EC 3.4.21.5. Sigma-Aldrich), Promod 278P (endopeptidase, EC 3.4.24.28, BISION, Sungnam, Korea), Multifect PR 14L (thermophilic-bacterial protease from Geobacillus stearothermophilus, EC 3.4.24.27, BISION), and Protex 6L (alkaline protease from Bacillus licheniformis, EC 3.4.21.65, BISION). Enzymes were added at 1:50 (enzyme: substrate) ratio, and then the mixture was incubated for 4 h under optimal conditions: Trypsin (pH 8.0, 40°C), Promod 278P (pH 6.5, 45°C), Multifect PR 14L (pH 8.0, 70°C), and Portex 6L (pH 7.0, 60°C). To inactivate the enzymes, the mixture was heated for 10 min at 100°C, and then centrifuged at 2,000×g for 20 min. The supernatant was lyophilized using a freeze-dryer. The OVA hydrolysates produced by Trypsin, Promod 278P, Multifect PR 14L, and Protex 6L were named as OHT, OHPM, OHMF, OHP, respectively, and the size of OVA hydrolysates was determined using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (15% gel).
The RAW 264.7 cell line, which is a murine macrophage, was purchased from the KCLB (Korean Cell Line Bank, Seoul, Korea). The medium used for the growth of RAW 264.7 cells was DMEM that contained 1% penicillin-streptomycin and 10% FBS (heat-inactivated). RAW 264.7 cells were grown at 37°C in a humidified 5% CO2 incubator (MCO-18AIC, Sanyo, Osaka, Japan) and were sub-cultured at 70%–80% confluence.
The MTT assay (Lee et al., 2017b) was used for determining the effects of OVA and OVA hydrolysates on cell viability of RAW 264.7 cells. After measuring NO concentration in the medium, an aliquot of the MTT solution (2.5 mg/mL) was added to each well and incubated for an additional 4 h. Supernatants were removed, and dimethyl sulfoxide was added to the wells to dissolve the formazan crystals. The absorbance of each well was then measured using a microplate reader (Emax, Molecular Devices, Sunnyvale, CA, USA) at 570 nm. Cell viability was calculated using the following equation:
RAW 264.7 cells (2×105 cells/well) were plated into a 96-well plate for 2 h, then 2 mg/mL of OVA and OVA hydrolysates were treated and incubated for 24 h. NO concentration was measured using the Griess reagent (Kim et al., 2018). One hundred μL of supernatant mixed with 100 μL of the Griess reagent and the mixture was incubated for 15 min at room temperature. The absorbance at 540 nm was recorded for each well, using a microplate reader. NO concentration was calculated using a standard curve made of sodium nitrate.
RAW 264.7 cells (2×105 cells/well) were plated into a 96-well plate and various concentrations (0.5, 1, or 2 mg/mL) of OVA hydrolysates were treated for 2 h. After that, the plate was further incubated with or without LPS (100 ng/mL) for 24 h. NO concentration was then measured and calculated using the Griess reagent as mentioned above.
RAW 264.7 cells (5×105 cells/well) were plated into a 6-well plate and various concentrations (0.5, 1, or 2 mg/mL) of OVA hydrolysates were added and incubated for 24 h. At the end of incubation, the samples in the plate were treated with LPS (0 or 100 ng/mL) and then incubated for 24 h. Total RNA was isolated using the RNA isolation kit according to the manufacturers' instructions and synthesized to cDNA using a synthesis kit. Synthesized cDNA was used in PCR analysis as template DNA. The primer sequences were shown as follows: iNOS (forward 5’-CCCTTCCGAAGTTTCTGGCAGCAGC-3’, reverse 5’-GGCTGTCAGAGCCTCGTGGCTTTGG-3’), and β-actin (forward 5’-GTGGGCCGCCCTAGGCACCAG-3’, reverse 5’-GGAGGAAGAGGATGCGGCAGT-3’). The RT-PCR reaction was conducted under the following reaction conditions: 95°C for 5 min (initiation), and followed by 25 cycles of 95°C for 30 s (denaturation), specified temperature of each primer for 30 s (annealing), 72°C for 45 s (extension), and followed by a final extension at 72°C for 10 min. The final products were then evaluated by 1.2% agarose gel electrophoresis with DNA SafeStain (Thermo Fisher Scientific). For quantitative real-time PCR analysis, SYBR reagent was used and the reaction was conducted under the following reaction conditions: 95°C for 2 min (initiation) followed by 40 cycles of 95°C for 5 s (denaturation), 60°C for 30 s (annealing/ extension). The amplified results were analyzed using the delta-delta Ct method and normalized to β-actin. The purity of PCR products was assessed by analyzing the melting curve.
RAW 264.7 cells (2×106 cells/well) were plated into a 6-well plate and OVA hydrolysates were treated and incubated for 24 h. After that, LPS was added to each well and further incubated for 45 min. RAW 264.7 cells were washed with PBS and lysed using the Radioimmunoprecipitation assay (RIPA) lysis and extraction buffer (Thermo Fisher Scientific) with protease/phosphatase inhibitor. Total protein concentration was measured by Lowry’s method. And the same concentration (25 μg) of proteins were separated by 12% SDS-PAGE gel and transferred to polyvinylidene fluoride membrane (PVDF) membranes. After blocking with 5% skim milk or 5% BSA dissolved in TBST (Tris-buffered saline with 1% Tween-20) for 1 h, membranes incubated with primary antibody iNOS, β-actin, total or phosphorous from of MAPKs (p38, JNK, and ERK) in 5% skim milk or 5% BSA overnight at 4°C. After washing 3 times with TBST, the membranes were incubated with secondary antibody (Goat anti-mouse IgG-HRP) for 2 h at room temperature. After washing, the protein bands were detected using a chemiluminescent ECL kit (Thermo Fisher Scientific).
SPSS 18.0 software (Chicago, IL, USA) was used for statistical analysis. Data were expressed as mean±SD. All data were taken from three independent experiments. For analysis between two samples, the Student's t-test was used. One-way analysis of variance (ANOVA) followed by Duncan’s multiple range test was used for the analysis between multiple samples. The significance of differences was reported at the level of p<0.05.
Results and Discussion
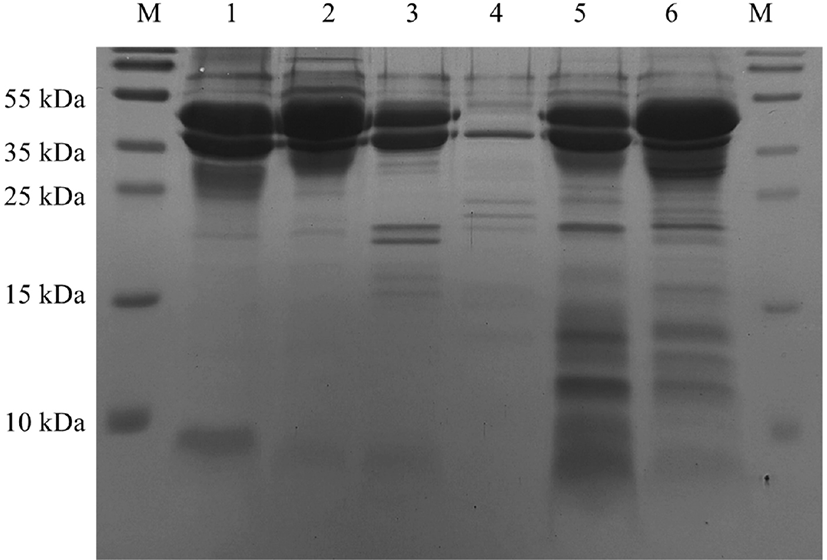
The OVA standard (lane 1) showed two thick bands near 45 kDa, and our OVA (lane 2) showed a similar band pattern (Fig. 1). Lanes 3–6 showed the results for the OVA hydrolysates prepared using Multifect 14L hydrolysate (OHMF), Promod 278P hydrolysate (OHPM), Protex 6L hydrolysate (OHPT), and trypsin (OHT), respectively. Although the same amounts of samples were loaded on lanes 3–6 (1 mg/mL), the density of the band observed around 45 kDa in Lane 4 was lighter than that of other lanes, indicating that Promod 278P hydrolyzed OVA better than other enzymes used. Also, lanes 3–6 showed different band patterns: lanes 5 and 6 showed more peptide bands than lanes 3 and 4 in the areas where the molecular weights were <25 kDa. This means that the four proteolytic enzymes produced different kinds of peptides from the OVA. Also, this is why each OVA hydrolysate exhibited different activities. Lee and Paik (2019) reported that the biological activity of protein hydrolysates and peptides were closely related to their amino acid sequence, length, and composition. Some researchers purified and identified some of the main peptides to determine their functional activity, but many others examined the functional activity of protein hydrolysates or mixtures of various peptides (Bhaskar et al., 2019; Lee et al., 2017a; Millán-Linares et al., 2014). In this study, we have investigated the anti-inflammatory activity of OVA hydrolysates, instead of individual peptides in the hydrolysates, in LPS-stimulated RAW 264.7 cells.
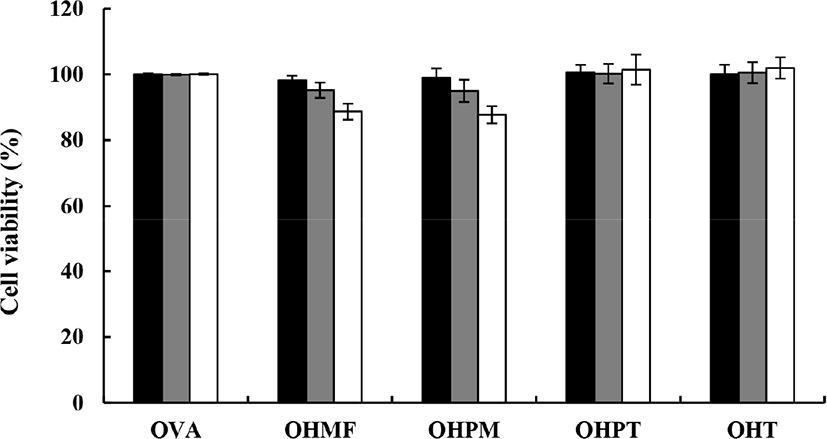
The effects of OVA and OVA hydrolysates on cell viability were evaluated using MTT assay to confirm that the changes of NO production by the LPS-stimulated RAW 264.7 cells were not the result of cytotoxicity of the hydrolysates. As shown in Fig. 2, the viability of the RAW 264.7 cells in all groups was above 90%, indicating that all concentrations (0.5–2 mg/mL) of OVA and OVA hydrolysates were not toxic to the RAW 264.7 cells. Thus, we could confirm that the changes of NO production in RAW 264.7 cells by the OVA and OVA hydrolysate treatments were not the result of cytotoxicity.
OVA has been widely used as an antigen in experimental animal models (including as an inhalant) and dietary allergy study (Mine and Yang, 2008). Some studies reported that OVA increased iNOS and COX-2 expression by activating NF-κB pathways in RAW 264.7 cells (Lee et al., 2011). However, this allergenicity could be decreased by heat, pH, and enzymatic hydrolysis because these parameters could alter protein structure (Ballmer-Weber et al., 2016; Duan et al., 2014).
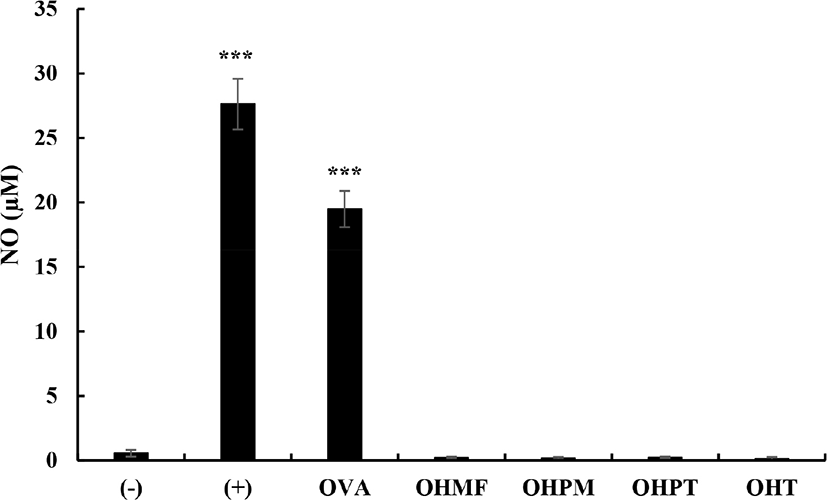
Thus, to confirm the inflammatory response to OVA (allergenicity) and its hydrolysates, RAW 264.7 cells were treated with OVA and OVA hydrolysates and the amount of NO produced was determined. As shown in Fig. 3, LPS (100 ng/mL) treatment increased the NO production level to 27.61±1.96 μM and addition of OVA to the LPS-stimulated RAW 264.7 cells increased the NO production level to 19.48±1.40 μM. However, the addition of OVA hydrolysate to the LPS-stimulated RAW 264.7 cells produced less than 1 μM of NO (OHMF, OHPM, OHPT, and OHT showed 0.20±0.08, 0.17±0.08, 0.22±0.07, and 0.13±0.13 μM, respectively), which were similar to the amount produced in the cells added with distilled water (negative control; 0.55±0.26 μM), indicating that OVA hydrolysates did not affect NO production.
As shown in Fig. 4, the positive control (LPS, 100 ng/mL) showed high production of NO (28.39 ±1.63 μM). OVA hydrolysates showed significant inhibitory effects on NO production. At a concentration of 2 mg/mL, OHMF inhibited NO production by 24.24±1.35 μM. The OHPT and OHT treatments showed dose-dependent inhibitions of NO production at 0.5, 1, or 2 mg/mL (OHPT: 25.17±1.46, 21.15±1.35, and 17.01±0.84 μM, OHT: 23.28±1.35, 19.46±1.07, and 13.99±2.22 μM, respectively).
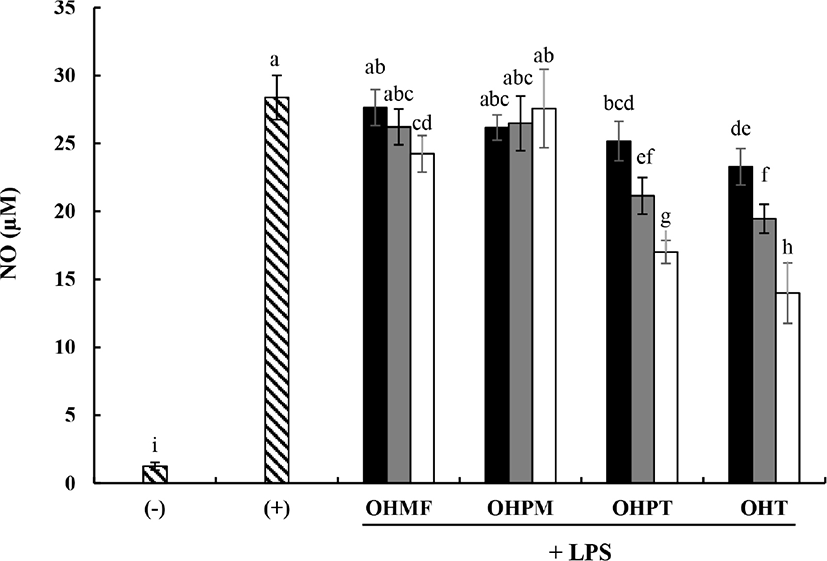
Under normal conditions, NO performs an important function in the inflammatory response (Kumar et al., 2017). When inflammation is induced, an appreciable level of NO is secreted by immune-activated macrophages at the site of inflammation. However, the overproduction of NO causes various complications as a result of chronic inflammation (Cho et al., 2011). Therefore, the inhibitory effect of OVA hydrolysates on NO production in LPS-stimulated RAW 264.7 cells is a relevant measure to releave the inflammatory activity (Kim and Kim, 2019). Several studies have suggested that the peptides in the hydrolysates prepared using proteolytic enzymes exerted anti-inflammatory activity by inhibiting NO production in the LPS-stimulated RAW 264.7 cells (Kim et al., 2013; Lai et al., 2011). As shown in the results, OHT showed the highest inhibitory effect on NO production in the LPS-stimulated RAW 264.7 cells. Therefore, OHT was selected for further analysis.
To understand the inhibition mechanism of NO production by OTH, the amount of iNOS and the expression of iNOS mRNA were evaluated using Western blotting and PCR analysis, respectively.
The inducible NO synthase is strongly linked to the regulation of NO production during the inflammatory responses. Hence, many researchers have tried to find new substances that can inhibit its enzyme production (Shanura Fernando et al., 2018). As shown in Fig. 5A, the production of iNOS was significantly decreased dose-dependently by OHT. The expression of iNOS mRNA (Fig. 5B) also significantly decreased when treated with OHT (p<0.05). These decreases, 23.34±3.56, 43.79±2.59, and 63.87±1.47% at 0.1, 0.5, and 2 mg/mL of OHT, respectively, also showed dose-dependence. The expression of iNOS was not affected when the RAW 264.7 cells were treated with OHT only.
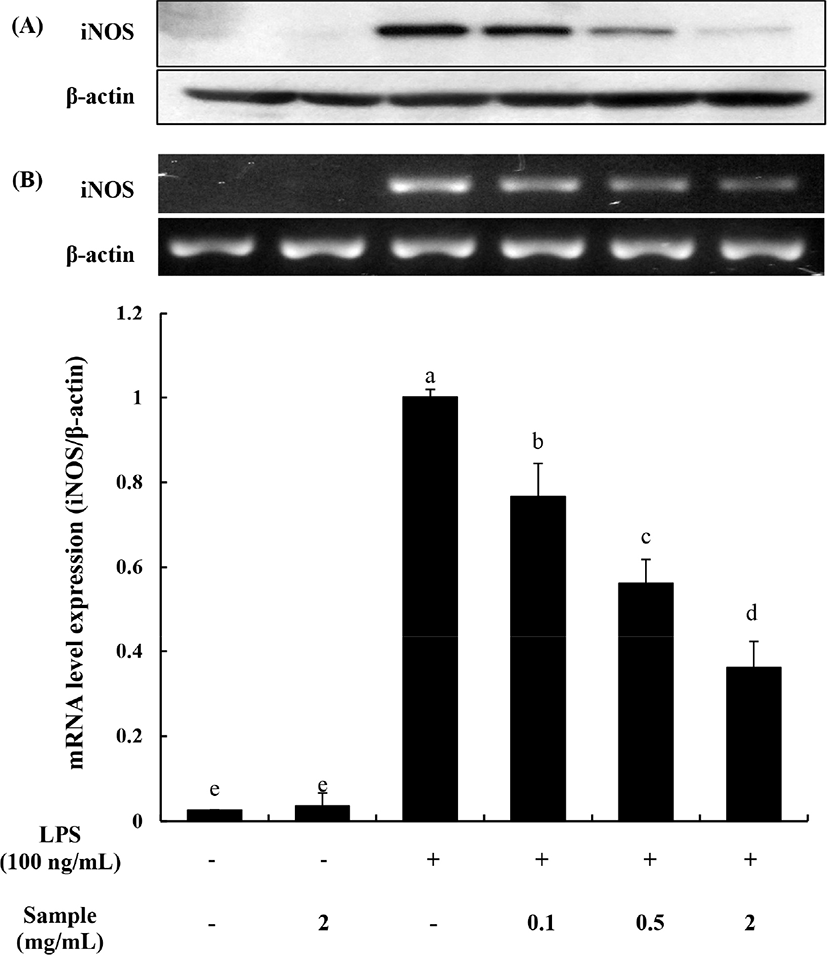
After stimulation, macrophages produced increased amount of iNOS, indicating that the pro-inflammatory response is directly related to the production of NO. The overexpression of iNOS enzyme produces excessive amount of NO and leads to the pathogenesis of various inflammatory diseases, septic shock, and cancer (Lai et al., 2011). Many studies reported that natural compounds including carbohydrates, protein, and flavonoids have anti-inflammatory activity by suppressing iNOS expression, resulting in the inhibition of NO production (Ham et al., 2015; Shanura Fernando et al., 2018; Sun et al., 2016). Thus, our results suggested that OHT inhibited NO production by inhibiting iNOS expression.
The Western blotting was performed to confirm whether the MAPK pathway is involved in the anti-inflammatory activity of OHT in LPS-stimulated RAW 264.7 cells. The JNK, ERK, and p38 pathway are part of the MAPK pathway whose phosphorylation increases their inflammatory activity. As shown in Fig. 6, the phosphorylation of JNK, ERK, and p38 in RAW 264.7 cells increased after the cells were treated with LPS (45 min). However, when RAW 264.7 cells were co-treated with LPS and OHT (2 mg/mL), OHT inhibited the phosphorylation of JNK and ERK but did not affect the phosphorylation of p38.
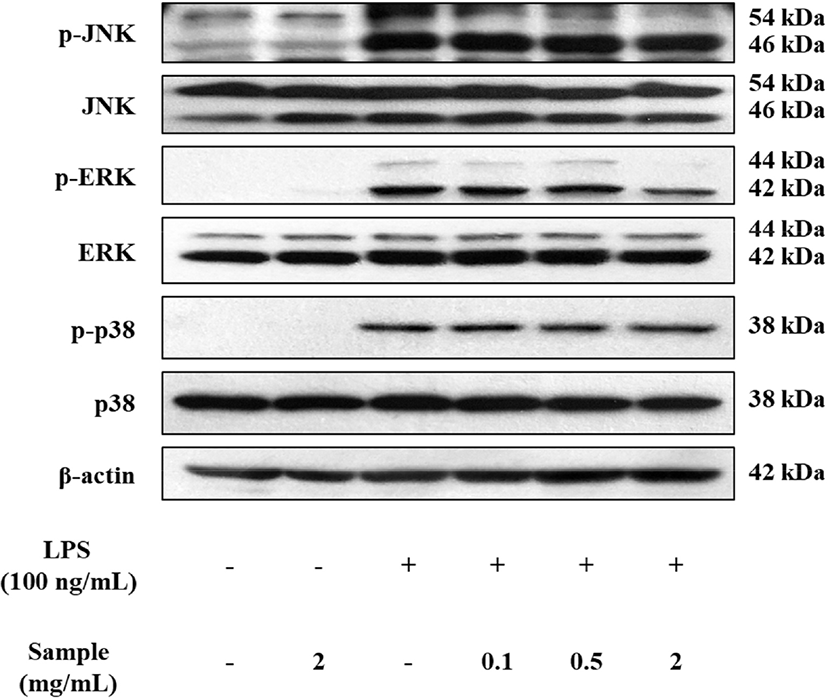
The MAPK pathway is known to regulate several cellular responses (e.g., cell survival, proliferation, and apoptosis) following exposure to various stimuli such as stress, shock, and pro-inflammatory cytokines (Pearson et al., 2001). Also, many studies indicated that the phosphorylation of MAPK was associated with the pro-inflammatory signaling, and up-regulate the production of IL-6, iNOS, and COX-2 in LPS-stimulated macrophages (Gao et al., 2012; Ham et al., 2015). Therefore, effective anti-inflammatory materials should show inhibitory activity on the phosphorylation of MAPK genes. Crebanine, isolated from Stephania venosa, inhibits the production of NO, iNOS, COX-2, and PGE2, and also inhibits pro-inflammatory cytokines including TNF-α and IL-6 by suppressing the MAPK signaling pathways (Intayoung et al., 2016). A study with sophocarpine, which is an alkaloid from Sophoraalopecuroides L., indicated that sophocarpine attenuated the phosphorylation of p38 and JNK, resulted in the inhibition of NO production (Gao et al., 2012), which was similar to our results.
Conclusion
This study demonstrated that OVA hydrolysate treated with trypsin (OHT) showed significant anti-inflammatory activity. The OHT inhibited NO production in the LPS-stimulated RAW 264.7 cells by suppressing the production of iNOS and the expression of iNOS mRNA. Also, it was confirmed that the anti-inflammatory activity of OHT was through the inhibition of JNK and ERK pathways. Therefore, the tryptic hydrolysates of OVA could be used as an anti-inflammatory agent in food products.