










Introduction
The unique flavor of meat is determined by specific metabolites such as amino acids, fatty acids, purines, and sugars, which vary depending on the type of muscle, species, sex, and feeding state (Jayasena et al., 2013; Lin et al., 2007; Piao et al., 2018). Meat flavor is formed during cooking via complex reactions including the Maillard reaction, thiamine degradation, and lipid oxidation, which are depending on the types of metabolites present (Jayasena et al., 2013; Lee et al., 2017). In addition, all post-slaughter conditions such as the packaging method, process temperature, and storage time can affect the metabolite contents and flavor of meat (Aliani et al., 2013; Zhu et al., 2001). To investigate the effects of various conditions on meat flavor, a fast and accurate analysis method for quantifying the metabolites is needed.
In general, each metabolite in a meat sample must be quantified using a specific technique. For example, free amino acids, sugars, and nucleotide-degraded compounds are analyzed by chromatography or spectroscopy with specific pre-treatments (Choe et al., 2010; Jayasena et al., 2015; Jung et al., 2013). Among the different instrumental analytical methods, nuclear magnetic resonance (NMR) spectroscopy provides several advantages compared to conventional methods including easy sample preparation, short running time, and the use of only one reference compound (Simmler et al., 2014). Thus, NMR spectroscopy has become commonly used in the field of quantitative analytical chemistry. Quantitative NMR (qNMR) has been applied to pure substance and complex natural samples in various fields such as pharmaceutical analysis, medical diagnosis, natural products analysis, and food science (Galo et al., 2015; Ramakrishnan and Luthria, 2017; Simmler et al., 2014). In addition, NMR spectroscopy for meat shows excellent reproducibility and accuracy compared to chromatographic analysis, and can quantitatively measure nearly all metabolites simultaneously (Gallo et al., 2015).
Despite these advantages, qualification and quantification conditions must be optimized before NMR analysis of meat metabolites. For example, the chemical characteristics and polarity of metabolites to be tested should be considered to ensure accurate analysis. Because of this, different extraction solutions are used depending on tissue type and metabolite polarity, which can be optimized before analysis (Lin et al., 2007). In addition, the extract must be stored under appropriate conditions including pH and salt concentration (≤0.15 M) for accurate quantitative analysis and to prevent adverse effects such as chemical shift or incorrect results in multivariate data analysis (Xiao et al., 2009).
Several previous studies have applied NMR to quantify meat metabolites including free amino acids and fatty acids and evaluated the characteristics of meat by multivariate data analysis (Graham et al., 2010; Jung et al., 2010; Siciliano et al., 2013). In these studies, an existing quantitative method was applied for different samples without additional verification procedures such as conventional HPLC (high-performance liquid chromatography) quantification. Additionally, they focused only on multivariate data analysis to evaluate the possibility of using NMR (Simmler et al., 2014). Few studies have optimized the analysis conditions for specifically qualifying and quantifying meat metabolites for further related study. Therefore, the objectives of this study were to develop an optimum qNMR method to qualify and quantify polar metabolites in chicken breast based on the extraction solution, reconstitution buffer, and NMR acquisition condition (pulse program). Then, the optimized method was tested for beef and pork.
Materials and Methods
DSS [3-(Trimethylsilyl)-1-propanesulfonic acid] sodium salt, L-aspartic acid, L-glutamic acid, creatine, deuterium oxide, deuterium oxide including TSP [3-(trimethylsilyl)propionic-2,2,3,3-d4 acid] sodium salt, HEPES [4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid], and mono- and di-phosphate sodium salt (anhydrous form) were purchased from Sigma-Aldrich (St. Louis, MO, USA). MOPS (3-Morpholinopropanesulfonic acid) was purchased from Amresco (Solon, OH, USA).
Chicken breast meat (Pectoralis major), beef (Semitendinosus), and pork (Longissimus thoracis) were purchased from a commercial market (Seoul, Korea). The meats were ground in a laboratory using a meat grinder (MG510, Kenwood Appliances Co., Ltd., Dongguan, Guangdong, China). Five grams of the ground meats were transferred into a 50 mL test tube and stored at −70°C with vacuum packaging. The meats in the frozen state were thawed at 4°C for 24 h before analysis.
To investigate the influence of different buffers on the NMR spectra, three extraction solutions [0.6 M PCA (perchloric acid), 10 mM phosphate buffer (pH 7.0), and methanol/water (1:1 ratio)] and three reconstitution buffers (20 mM HEPES, MOPS, and phosphate buffer) were prepared, respectively. All solutions were prepared with deionized distilled water (DDW). After decision of the optimum conditions for the extraction solution and reconstitution buffer using chicken breast meat, the beef and pork were tested to confirm the method whether it can be utilized for samples from different species.
The thawed ground chicken breast meat (5 g) was extracted with 20 mL of different extraction buffers using a homogenizer (T25 basic, Ika Co., KG, Staufen, Germany) for 2 min at 1,871×g. The extraction buffers were 0.6 M PCA, 10 mM phosphate buffer (pH 7.0), and methanol/water (1:1 ratio). The homogenate was centrifuged (Continent 512R, Hanil Co., Ltd., Gyeyang-gu, Incheon, Korea) at 3,500×g for 20 min. Each supernatant was transferred into new test tube and titrated to pH 7.0 with 1 M and 6 M sodium hydroxide solution, and then centrifuged again (Continent 512R, Hanil Co., Ltd.) under the same conditions. Each supernatant was filtered (Whatman No. 1, Whatman PLC., Brentford, Middx, UK) and lyophilized (Freezer dryer 18, Labco Corp. Kansas City, MO, USA) prior to NMR analysis.
The dry mass of the lyophilized extracts was reconstituted using 3 different buffers (20 mM HEPES, MOPS, and phosphate buffer) for comparison. HEPES and MOPS buffers were titrated with sodium hydroxide and phosphate buffer with sodium mono- and di-phosphate to adjust the pH to 7.0. All reconstitution buffers were prepared with D2O containing 1 mM TSP for quantification of metabolites in the meat sample.
1D 1H NMR spectra were acquired by ICON-NMR automation (Bruker Biospin GmbH, Rheinstetten, Baden-Württemberg, Germany). Lock, tune, and shimming were performed automatically. After acquisition, spectra were processed with Topspin 3.5pl7 (Bruker Biospin GmbH). Phase correction and integration of the peak of interest were performed manually.
A modified standard Bruker pulse program (noesypr1d and zg30) was used for quantification. Each spectrum was recorded in D2O on a Bruker 600 MHz Cryo-NMR spectrometer (Bruker GmbH). Spectra were obtained at the 1H frequency of 600.130 MHz applying a modified standard noesypr1d (recycle delay of 15 s) or zg30 (recycle delay of 1 s) pulse sequence, with the lock on the deuterium resonance of the solvent. The experimental parameters were as follows: sweep width of 7,812.500 Hz, 128 K data points. In noesypr1d and zg30, two prior dummy scans were applied for each spectrum and 64 and 128 scans were recorded, respectively. Spectral processing was carried out using Topspin 3.5p7 (Bruker GmbH). The chemical shifts (δ) were referenced to the TSP resonance or DSS resonance. Baseline correction was performed manually. The major difference between noesypr1d and zg30 is the water suppression function in noesypr1d (Fig. S1 of supplementary material).
Peaks (no or little overlap) of the metabolites were identified based on the chemical shift according to Human Metabolome Database (www.hmdb.ca) and Chenomx NMR suite 7.1 (Chenomx, Inc. Edmonton, AB, Canada). Each metabolite was calculated using 1 mM TSP as an internal standard. The concentration of metabolites was quantified using the following equation.
The reconstituted samples for NMR analysis were diluted 10 times for amino acid quantifications (Schwarz et al., 2005). The samples were filtered through a membrane filter (0.2 μm) into a glass vial and injected into an HPLC system (Ultimate 3000, Thermo Fisher Scientific, Inc., Waltham, MA, USA). In the reaction chamber, after injecting 5 μL borate buffer (PN 5061-3339, Agilent Technologies, Santa Clara, CA, USA), each 1 μL sample, o-phthalaldehyde reagent (PN 5061-3335, Agilent), and 9-fluorenylmethyl chloroformate solution (PN5061-3337, Agilent) were reacted and diluted with 32 μL deionized water DDW. Next, the solution (0.5 μL) was injected to the column with an elution time of 30 min. A VDSpher 100 C18-E column (4.6×150 mm, 3.5 μm, VDS Optilab Chromatographie Technik GmbH, Würzburg, Germany) was used with 40 mM sodium phosphate, dibasic (pH 7.8) and DDW/acetonitrile/methanol (10:45:45 v/v %) as the mobile phase with a slightly modified gradient (Henderson et al., 2014); the flow rate was 1.5 mL/min. The column temperature was maintained at 40? and detection was monitored at wavelengths of 266 and 340 nm. Each amino acid content was calculated from the area of each peak using standard curves obtained using amino acid standards (PN 5061-3330 and 5062-2478, Agilent).
Statistical analysis was performed using the procedure of the general linear model. Significance of differences among mean values were determined by Student-Neuman-Keul’s test using SAS software with a confidence level of p<0.05 (SAS 9.4, SAS Institute Inc., Cary, NC, USA). All the experimental procedures were conducted in triplicate.
Results and Discussion
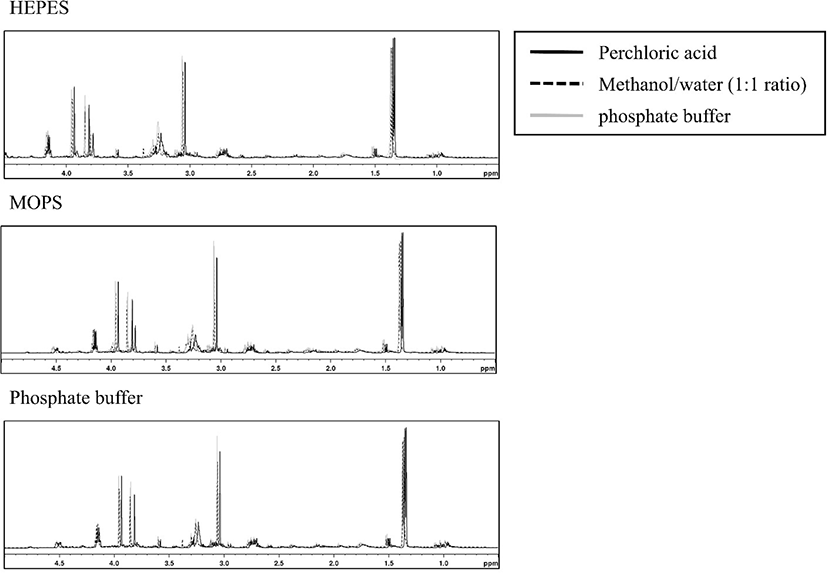
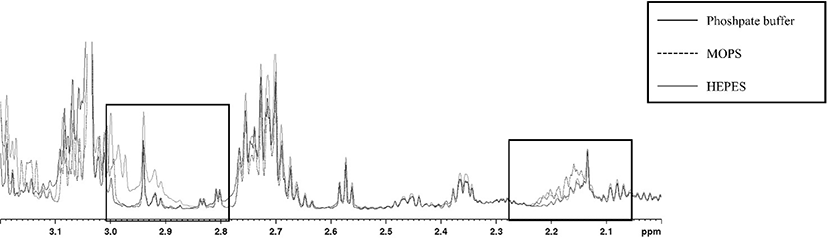
For extracting polar metabolites, three different solutions (methanol/water, phosphate buffer, and perchloric acid) were selected, which are commonly used, in the present study (Dietmair et al., 2010; Lin et al., 2007; Maharjan and Ferenci, 2003; Römisch-Margl et al., 2012). Among the combinations of extraction solutions and reconstitution buffers used to optimize the buffer conditions, the extracts of chicken breast meat using methanol/water (1:1 ratio) showed an unflattened baseline, regardless of the reconstitution buffers tested. Similarly, Lin et al. (2007) reported that methanol/water (1:1 ratio) extraction showed high-yield and was fast and easy to operate, but exhibited low reproducibility. Phosphate buffer extraction showed results similar to those with methanol/water (1:1 ratio), except that HEPES was used as a reconstitution buffer. Phosphate buffer is mainly used to extract heme pigments such as myoglobin and hemoglobin (Warris, 1979) and these extracts are not used to analyze polar metabolites. In addition, extraction using acidic solution may be advantageous for extracting more amounts of amino acids (Aristoy and Toldra, 1991). In contrast, extraction with perchloric acid using MOPS and 20 mM phosphate buffer as reconstitution buffer showed a flattened baseline (Fig. 1). Wishart (2008) reported that organic buffers (MOPS and HEPES) should be avoided in the complex mixture sample for NMR analysis because overlap of target peaks can occur (Fig. 2). DSS as an internal standard that generates overlap downfield and may disappear, particularly in phosphate buffer extract (Fig. S1 of the supplementary material). Nowick et al. (2003) reported that DSS can interact with cationic peptides and convert to different compounds. Thus, DSS was not an appropriate internal standard for the present study and we used TSP to quantify metabolites in chicken meat samples. Based on our results, extraction with perchloric acid and reconstitution in phosphate buffer was the best preparation condition for qNMR to quantify the metabolites in chicken breast.
Two NMR pulse programs (zg30 and noesypr1d) were compared to optimize the method for quantifying polar metabolites in chicken breast. Pulse program noesypr1d, which included a water suppression process, led to an increase in the delay time to obtain the quantitative area of the NMR signal (Bharti and Roy, 2012). Because zg30 involves no water suppression process, NMR spectra were acquired more rapidly (approximately 10 min) than with noesypr1d. For qNMR analysis, whether signal intensity is quantitatively obtained without water suppression process because of different chemical characteristics of each sample should be tested prior to application (Bharti and Roy, 2012; Krssák et al., 2004).
A previous study reported that a delay time with at least 5 T1 relaxations, the time for an excited state to return to the ground state in NMR analysis, is required to generate a sufficiently intense peak area for quantification (Saito et al., 2004). The delay times of both noesypr1d (over 15 s) and zg30 (1 s), limit of detection, and linearity were evaluated before qNMR analysis using a standard mixture (Table S1, S2, and Fig. S3 of the supplementary material). Water suppression normally provides maximum signal intensity of a sample by removing high water signals from the spectrum (Bharti and Roy, 2012). Both neutral pH and a water suppression process can improve signal/noise ratios and spectra stability (Araníbar et al., 2006). In the present study, qNMR analysis of the metabolites in chicken breast using the zg30 pulse program was less time-consuming and showed no difference in quantification capability.
Both NMR pulse programs (zg30 and noesypr1d) were performed to quantify metabolites in chicken breast extracts. To verify the accuracy of the developed qNMR method, the results of amino acid contents from HPLC analysis were compared. All analyses showed relative standard deviation (RSD) values less than 5%, regardless of the HPLC and different NMR pulse program, demonstrating good repeatability (Saranadasa, 2000). Especially, leucine and isoleucine showed higher concentration in NMR than HPLC analysis (p<0.05). The pulse program of zg30 showed a lower RSD than noesypr1d. Moreover, noesypr1d (water suppression) revealed an incorrect tyrosine concentration compared to the results obtained by HPLC. Tyrosine showed longer T1 relaxation time (5.13 s) than other target metabolites (Table S3 of the supplementary material). Thus, the delay time (15 s) of noesypr1d was not enough to obtain the signal intensity for quantification of tyrosine (Saito et al., 2004). However, the concentration of tyrosine by NMR analysis using zg30 pulse program was not different from that by HPLC. Mckay (2011) reported that any modified parameter in the analytical process, such as water suppression, may adversely affect the accuracy of NMR analysis. Our results showed that the zg30 pulse program performed equally well to HPLC analysis for free amino acid measurement. Among the tested amino acids, leucine, isoleucine, alanine, tyrosine, and phenylalanine showed better repeatability using NMR with the zg30 pulse program. However, valine, glutamic acid, aspartic acid, and glycine showed better repeatability in HPLC analysis based on the RSD (%). This wide variation of qNMR for several free amino acids may be because of the slight overlap in the spectra. Overall, we confirmed that qNMR analysis using an internal standard (TSP) is accurate for quantifying free amino acids (Table 1). NMR acquisition using the zg30 pulse program (without water suppression) can analyze metabolites more appropriate than noesypr1d because of the accuracy and its shorter analysis time.
All metabolites from chicken breast which could be qualified and quantified using the optimized conditions are represented in Fig. 3. Lactate and creatine were notably observed as metabolites in chicken breast meat extracted by perchloric acid. As observed in beef extracts (Graham et al., 2010), nucleotides (inosine-5′-monophosphate, hypoxanthine), dipeptides (creatine, anserine), and other metabolites were confirmed by peak multiplicity and the shape and region of peaks in the present study. However, the chemical shifts of metabolites were slightly varied (Graham et al., 2010; Table 1). Some metabolites qualified by Graham et al. (2010) were not observed in the present NMR spectra, which may be because of differences in the chemical environment of the samples, such as the metabolite ratio and concentration or ionic strength of the extract, which may lead to different chemical shifts in the NMR spectrum (Govindaraju et al., 2000).
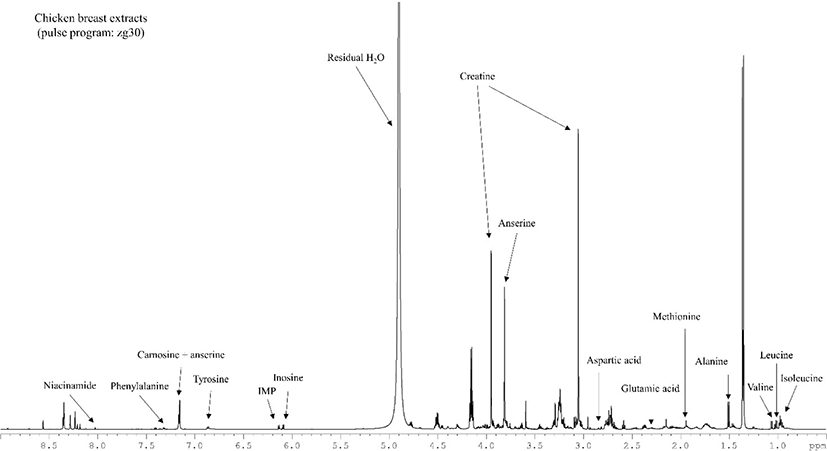
In contrast, 1D 1H qNMR studies have reported chronic overlap issues with different samples including bio-fluid, foods, or natural compounds. In the present study, we also observed peak overlaps in asparagine, serine, glutamine, histidine, threonine, taurine, and tryptophan, which may be a limitation of this method. In this regard, two-dimensional qNMR analysis is recommended to overcome this problem by separating each metabolite peak from the peak cluster (Martineau et al., 2012). Therefore, further development in 2D NMR approaches may help to detect metabolites in meat samples including several amino acids, dipeptides, and nucleotides that cannot be precisely quantified by 1D qNMR.
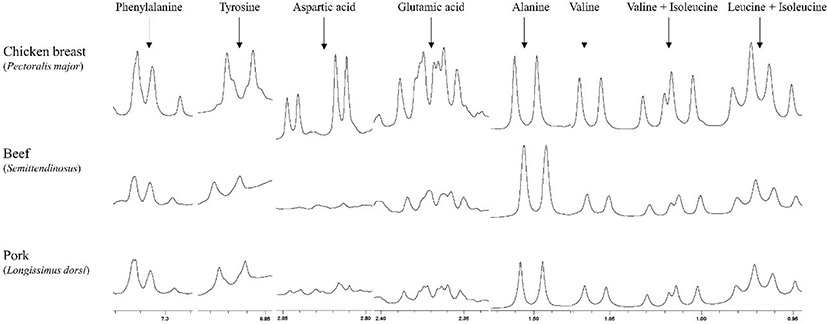
The optimized qNMR method using chicken breast was applied for beef and pork. From the method, the concentrations of free amino acids in beef and pork presented relatively lower than chicken breast (Table 2). When we observed spectra, alanine, leucine, isoleucine, glycine, glutamic acid, and phenylalanine were clearly qualified and quantified (Fig. 4). Aspartic acid of both beef and pork presented abnormal shapes of peak with partial overlap when compared with chicken meat but can be quantified. It could be one of sample-specific response among meats. The quantified free amino acid contents of pork and beef were similar to the data from the report of Cornet and Bousset (1999) and Feidt et al. (1996), respectively. The higher amount of free amino acids in chicken than beef and pork could be explained by fast postmortem metabolism rates (Schreurs, 2000).
Conclusion
To qualify and quantify the polar metabolites in chicken breast, extraction with perchloric acid and reconstitution with phosphate buffer showed the best results for qNMR analysis. In addition, spectra acquisition with zg30 without water suppression can greatly reduce the analysis time without compromising accuracy. Compared to chromatography, 1D 1H qNMR is an alternative analytical method that shows high-sensitivity, short running time, and good accuracy. Moreover, this optimized analysis method can be applied to beef and pork easily. Further studies should investigate a larger number of metabolites in meat for simultaneous quantification via advance techniques such as 2D hetero- and homo-nuclear qNMR analysis.