









Introduction
Barbiturates are anaesthetic and sleep-inducing agents that act through the central nervous system. They are widely used for general anaesthesia and the treatment of epilepsy (Coupey et al., 1997). However, they exhibit potentially dangerous side effects, including high blood pressures (Beaver et al., 1977), coma, shock and respiratory and heart problems (Effendi et al., 1974; Fregosi et al., 2004). Hence, the use of barbiturates in humans and animals is prohibited in many countries (Fan et al., 2012; Martin et al., 2016). However, some barbiturates are still used in animal feed and drinking water (Locket et al., 2010). Their consumption by dairy cows results in the presence of barbiturate residues in raw milk. Therefore, it is necessary to determinate the concentrations of barbiturates in raw milk to avoid subsequent hazards to human health.
Methods for the determination of one or more barbiturates have been published previously. These include capillary electrophoresis (CE) (Fan et al., 2012), gas chromatography (GC) (Johnson et al., 2010), liquid chromatography (LC) (Garcíaborregón et al., 2000) and mass-spectroscopy-coupled GC and LC (GC-MS and LC-MS, respectively) (Li et al., 2010; Shendy et al., 2016; Sun et al., 2017; Tong et al., 2015). In recent years, LC-MS has been frequently applied to the determination of veterinary drug residues because of its high sensitivity, specificity and reliability. For example, Sørensen (Sørensen et al., 2011) developed an LC-ESI-MS/MS method to simultaneously detect 15 veterinary drug residues, including five barbiturates. The analysis had a total runtime of 26 min, and the limits of detection (LODs) of the barbiturates were 72-320 µg/L. Arbeláez (Arbeláez et al., 2015) used an LC-MS/MS method for the simultaneous screening, identification and quantitative determination of eight hypnotic drugs, including four barbiturates, in river water and wastewater; the LODs were in the range of 2.5-50.0 ng/L. Lee (Lee et al., 2013) developed a high-throughput LC-MS/MS method for the determination of eight barbiturates in human plasma. Quantification was performed via multiple reaction monitoring (MRM) with negative-ion atmospheric pressure chemical ionisation (APCI). The total runtime was 3 min; an off-line extraction procedure was not required. The LODs ranged from 1.0 to 10.0 ng/mL. In addition, these methods have been applied to biological matrices, such as urine, blood, plasma, serum, feed and animal tissues. However, the determination of barbiturates in raw milk has not been previously reported in detail. Raw milk contains abundant proteins and fats, and its composition is influenced by numerous factors and may change unpredictably (Petit et al., 2001; Petit et al., 2004). Therefore, the analytical methods applied to other biological matrices may not be appropriate for the analysis of drug residues in raw milk.
The purpose of the present study was to develop a rapid, sensitive and specific method for the simultaneous determination of four commonly used barbiturates (phenobarbital, pentobarbital, amobarbital and secobarbital) in raw milk. The developed method includes the extraction, purification and qualitative and quantitative analyses of the analytes.
Materials and methods
Stock standard solutions (0.0998-0.1006 mg/mL) of phenobarbital, pentobarbital, amobarbital and secobarbital were obtained from Dr. Ehrenstorfer (Germany). Structures shown in Fig. 1.
Methanol, acetonitrile, hexane and ethyl acetate were of chromatographic purity, and purchased from either Thermo Scientific (USA) or Merck (Germany). Ultrapure water was obtained using a Milli-Q system from Millipore (USA). The glacial acetic acid and sodium chloride (analytical grade) were purchased from China National Pharmaceutical Group Co. (China).
Raw milk samples, including 50 real raw milk samples, were obtained from several local producing farms in Shanghai and frozen at −20℃ for 24 h without any additional treatment. The milk samples were thawed, homogenised and kept at room temperature before use.
For the optimisation of extraction and purification procedures, 10.0 g of raw milk (blank sample) was weighed in a plastic centrifuge tube, and 400 µL of the working standard mixture was added to achieve a final concentration of 40 ng/g milk. The QC samples were prepared by spiking 100, 200 or 400 µL of the working standard mixture into blank raw milk samples to achieve final concentrations of 10, 20 or 40 ng/g milk, respectively. Furthermore, all of the spiked samples were left to stand for 10 min to ensure the homogeneity of the samples.
The equipment used for sample extraction and purification included a SORVALL ST 16R centrifuge (Thermo), a rotary evaporator (RV 10 Basic, IKA, China), a nitrogen concentration instrument (ANPEL, China) and ultrapure water production equipment (Millipore, USA). LC-ESI-MS/MS analysis was performed using a 1290 Infinity series liquid chromatograph (Agilent Technologies, Germany) with an ACQUITY UPLC CSH C18 HPLC column (2.1 × 100 mm; 1.7-µm particle size) (Waters, USA) and a 6470 Triple Quad series mass spectrometer (Agilent).
The separations of the four target barbiturates were performed on the LC instrument using linear gradient elution. The mobile phase consisted of water and acetonitrile. The gradient was programmed as shown in Table 1. The total analysis time was 10 min. The flow rate was set at 0.3 mL/min and the injection volume was 5.0 µL. The temperature of the column was maintained at 25℃.
Quantitative and qualitative analyses were performed by LC-MS/MS with an electrospray ionisation (ESI) source operated in negative-ion mode. LC-MS/MS was operated in MRM mode to quantify the target barbiturates using the target ion pairs and relative conditions shown in Table 2.
Note: a = quantitative ion
Each barbiturate (1.0 mL) was transferred into a 10-mL volumetric flask. The solution was made up to the mark with methanol to produce an intermediate solution with a concentration of 10.0 µg/mL. To produce the working standard mixture (1.0 µg/mL), each intermediate solution (1.0 mL) was transferred into the same volumetric flask and then made up to the mark with methanol. The working solution mixture was freshly prepared on each day of analysis. To eliminate interference from the matrix, calibration standards (10, 50, 100, 200, 500 and 1000 ng/mL) were prepared using processed blank milk solution.
For each milk sample, 10.0 g of raw milk was pipetted into a 50-mL plastic centrifuge tube, to which 20.0 mL of 0.1% (v/v) acetic acid-acetonitrile was added. The sample was subjected to ultrasonication for 30 min. Subsequently, 5.0 g of sodium chloride was added to the tube and vortex oscillation was applied for 1.0 min, followed by centrifugation at 8000 rpm for 5 min. A 10.0-mL fraction of the acetonitrile layer was then transferred into a glass test tube, and then evaporated to dryness under a nitrogen stream. The dry residue was re-dissolved in 2.0 mL of 30% (v/v) acetonitrile-water solution.
Purification was carried out using a solid phase extraction (SPE) cartridge (ODS C18, 6 mL, 150 mg) (BESEP, China), which was preconditioned with methanol and 30% acetonitrile-water solution. The processed milk sample solution was then passed through the cartridge, washed with water and completely dried under vacuum. The analytes were eluted with different solvents (individual or combination of hexane, ethyl acetate and dichloromethane), and the eluates were evaporated to dryness under a nitrogen stream. Finally, the residues were reconstituted with 1.0 mL of 30% acetonitrile-water solution, filtered through a 0.22-μm organic membrane and then subjected to LC-ESI-MS/MS analysis.
Results and Discussion
Acetonitrile and ethyl acetate were used for the extraction of barbiturates. The acetonitrile was found to be superior to ethyl acetate at precipitating proteins (Hidvégi et al., 2014; Osnes et al., 2009). Therefore, it was used as the extraction reagent for the rest of the experiments. Furthermore, barbiturates are acidic and exhibit changes in their molecular morphologies under acidic conditions. Adding glacial acetic acid enhances the extraction of barbiturates by organic solvents. Thus, 0.1% glacial acetic acid was added to each sample to promote protein precipitation (Song et al., 2015).
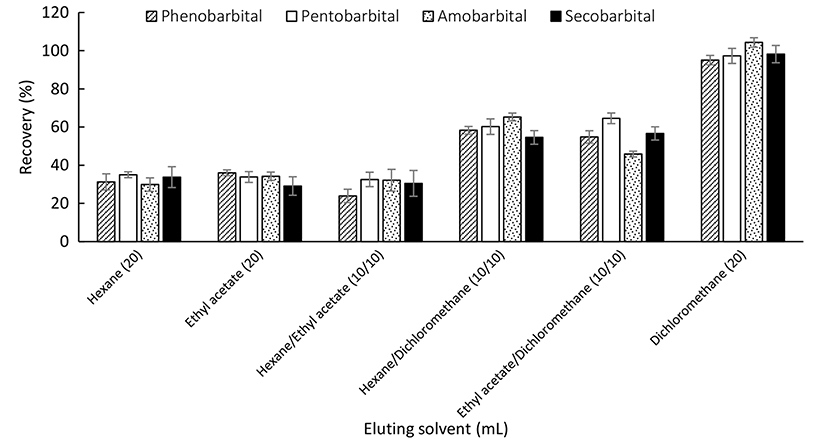
SPE cartridges were used to remove fat and other impurities from raw milk for it can significantly decrease the matrix effect (Chambers et al., 2007). The eluent was optimised for the purification of 40 ng/g milk standards on an SPE column (6 mL, 150 mg). Different proportions and volumes of hexane, ethyl acetate and dichloromethane were analysed. The results (Fig. 2) show that the elution ability of dichloromethane was much more efficient than others with the same volume and no significant change of recoveries was found with the increase of eluent volumes (date not shown), while the longer concentration time was needed. Finally, the barbiturate compounds can be almost totally eluted with greater recoveries when 20 mL dichloromethane was used.
It is difficult to construct an LC program capable of significantly separating the four target compounds because of their structural similarities. For instance, pentobarbital and amobarbital are isomers, which differ only in the position of a methyl group. To achieve adequate separation, the UPLC CSH C18 column was chosen because it exhibited acceptable separation of pentobarbital and amobarbital (Fig. 3(a)).
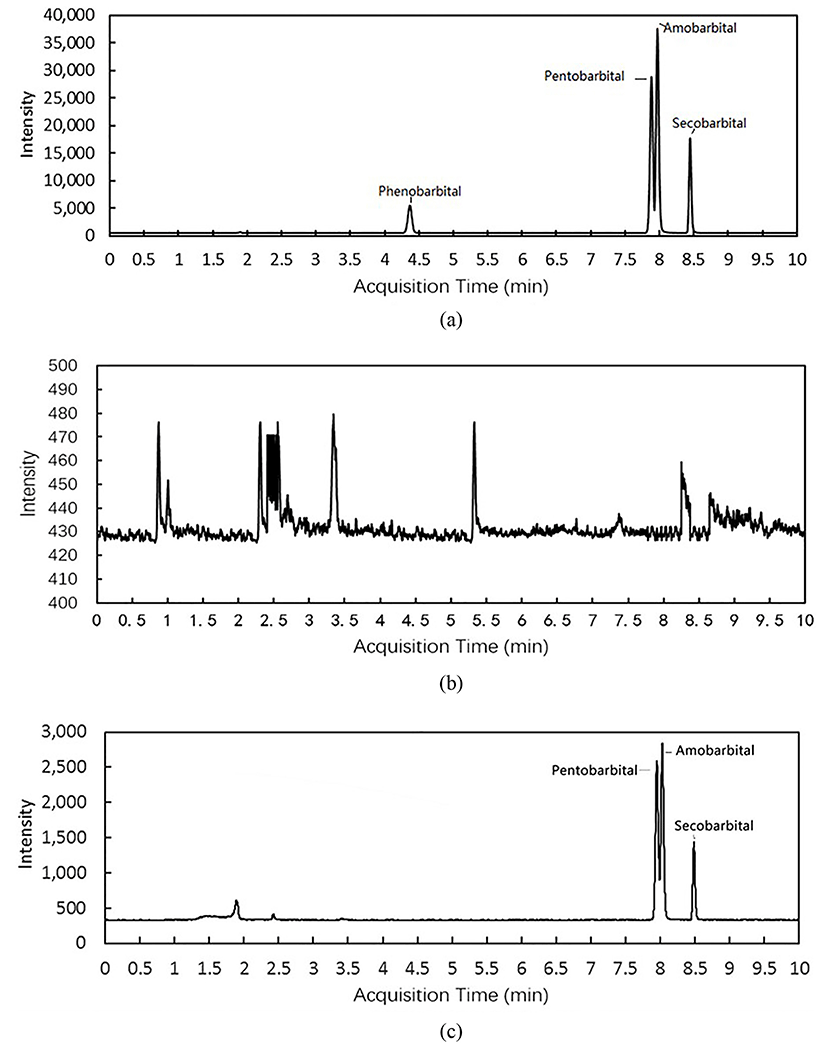
Optimising the mobile phase was crucial to achieve sufficient separation of analytes. The mobile phase may also influence the ionisation for MS analysis. Various individual and combinations of acetonitrile, methanol and water were investigated for use as the mobile phase. Acetonitrile was chosen as the organic solvent because its use resulted in greater peak sharpness and peak intensity (data not shown) than those obtained with methanol. Furthermore, acid was not added to the mobile phase because acidic conditions diminished the response of negative-ion ESI (Holcapek et al., 2004; Wu et al., 2004). The gradient elution programme developed in this study resulted in high peak symmetry and acceptable retention times for the barbiturates. A flow rate of 0.3 mL/min produced good peak shapes and permitted an LC runtime of 10.0 min. Figure 3a shows a chromatogram of the four target barbiturate standards (200 µg/mL of each standard).
Based on previous reports, negative-ion ESI was chosen as the MS ionisation method because it resulted in stronger barbiturate peak intensities than positive-ion ESI (Song et al., 2015).
The method was validated for selectivity, linearity, precision, accuracy, recovery and stability. The validation runs were conducted on three consecutive days. Each validation run included a single set of six calibration standards and six replicates of low-, medium- and high-concentration QC raw milk samples.
The selectivity of the method was determined by comparing the analyses of blank milk samples with those of blank milk samples spiked with barbiturates. No interference was observed at the retention times of the four target barbiturates.
Stability testing was conducted using six replicates of raw milk samples at three spiking concentrations. The samples were exposed to different conditions before analysis, including storage at room temperature for 24 h (short-term) or at 4℃ for 2 wk (long-term). The differences between the analyses were analysed using significance testing (Student’s T-test). The results indicated that all four of the barbiturates spiked into blank raw milk samples were stable under the conditions tested.
The linearity of the target compound peak area versus the calculated concentration was verified in raw milk using a 1/x2 weighted linear regression. And the linearity was conducted by external calibration, for it is a better way using the specific standard barbiturates to quantitative themselves. The correlation coefficient (r2) was > 0.99 for all of the target compounds in raw milk at concentrations of 0 to 1000 ng mL-1. Hence, the method exhibited good linearity. The LODs and calibration parameters for the target compounds in raw milk are shown in Table 3. The LOD was 5 ng/mL and the LOQ was 10 ng/mL for all four of the barbiturates. This is comparable with LODs obtained in previously reported studies (Lee et al., 2013), indicating that the proposed method is suitable for trace detection.
The recovery was determined by comparing the peak areas of raw milk standards spiked after extraction with the peak areas of post-extraction raw milk blank samples spiked before extraction with equivalent concentrations using six replicates. The accuracy was determined by comparing the known QC concentrations with corresponding concentrations fitted from calibration curves, six replicates as well. The precision was determined by calculating the relative standard deviation of three QC samples with different analyte concentrations, either in a single run (intra-assay precision) or between runs conducted on three consecutive days (inter-assay precision) (Zhang et al., 2016). Table 4 shows the precision, recovery and accuracy for each of the target barbiturates. The recoveries ranged from 85.0 to 113.5%, with intra-assay RSDs from 1.1% to 9.8% and inter-assay RSDs from 3.9% to 7.3%. These results are statistically acceptable.
Note: a = quantitative ion
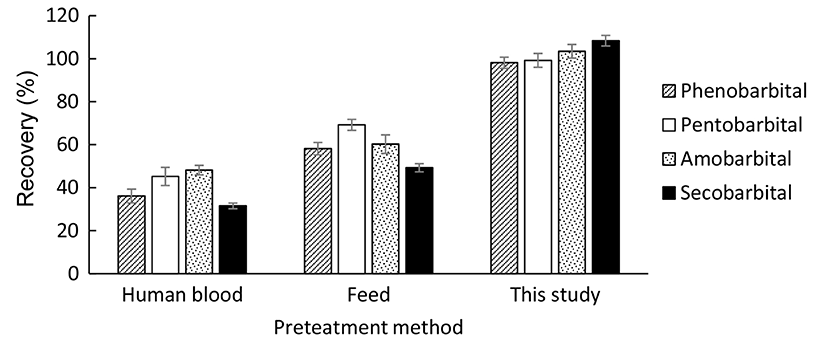
To verify the validity of the proposed method, pretreatment methods reported to extract barbiturates from human blood and feed were compared with the pretreatment method used in the present study. The same LC-MS/MS method was used for all of the analyses. The results were evaluated based on the recoveries of the four target barbiturates from 40 ng/g milk samples. As shown in Fig. 4, the recoveries obtained using the extraction methods developed for human blood (Zhang et al., 2016) and feed (Zhang et al., 2011) were significantly lower than the recoveries obtained using the method used in the present study, which meant the method developed in this paper was more suitable for determination of barbiturates in raw milk.
Fifty blind raw milk samples were obtained from local farms and analysed using the proposed method. Two of them were determined to be positive for barbiturates, and were retested to confirm the results (Table 5). Fig. 3(b) and 3(c) shows the chromatograms of blank sample and one positive sample. These results demonstrate that the proposed method is suitable for the determination of barbiturate residues in raw milk.
| Sample No. | Concentration (ng/g milk) | |||
|---|---|---|---|---|
|
|
||||
| Phenobarbital | Pentobarbital | Amobarbital | Secobarbital | |
| 16 | n.d. | 10.8 | 12.5 | 7.4 |
| 27 | 8.0 | n.d. | n.d. | n.d. |
Note: n.d. = not detected
Conclusion
A simple, sensitive and specific LC-MS/MS method was developed for the simultaneous determination of phenobarbital, pentobarbital, amobarbital and secobarbital in raw milk. Extraction was conducted using 0.1% (v/v) glacial acetic acid-acetonitrile, and an SPE column was used to remove fats and other impurities for decreasing of the matrix effect. High sample recoveries were obtained. Moreover, the validated results showed that the proposed method achieved good sensitivity, selectivity, accuracy and precision for the target analytes with matrix-matched calibration. The proposed method was successfully applied to the determination of target barbiturate residues in 50 raw milk samples, thereby demonstrating its suitability for monitoring and controlling veterinary drug residues in food matrices.