










Introduction
Milk is nature’s most nutritious food comprising of appreciable amount of essential nutrients and micronutrients, such as proteins, lipids, carbohydrates, vitamins and enzymes, and plays an irreplaceable role and position in the human diet (Abriouel et al., 2008; Thorning et al., 2016). Because of its nutritional properties, milk is also a good culture medium for a variety of spoilage and potentially pathogenic microorganisms which are harmful to human health. With the rapid development of China economy, the increasing demand for dairy products, milk safety problems become the focus of social attention (Gabriels et al., 2015). The quality and safety of raw milk, which is the upstream of dairy supply chain, are the main factors that limit the sustainable and healthy development of dairy industry (Coorevits et al., 2008). The quality of milk is affected by many factors: health status of cows, milk handling and hygiene of milking. The pathogenic microorganisms in raw milk are mainly bacteria, its spoilage causes significant economic losses for the food industry also can affect the health of consumers and even lead to death while lowering the quality of milk (De Silva et al., 2016; Salovuo et al., 2005).
A growing number of scientific studies indicated that the contamination of raw milk before milking was very low, mainly during milking and milk storage. Milk is considered to be sterile when secreted from a healthy udder, after which numerous contamination sources increase its bacterial load (Vacheyrou et al., 2011). The raw milk secreted by healthy cows is in a relatively sterile state, but the raw milk is inevitably contaminated by microorganisms at every procedures down the production chain, such as being squeezed out and transportation to dairy processing plants (Sørensen et al., 2016).
The research on the source of raw milk microorganisms has been a hot spot in foreign countries in recent years. In China, the research focuses on the studies of pathogenic bacteria but there is little research on the microorganism pollution source of raw milk (Garedew et al., 2012; Marjan et al., 2014). In order to control the microbial contamination and milk safety risk in raw milk, bacteria population dwelling in the production chain and environment of dairy industry in China should be strictly regulated and controlled. Therefore, it is important to characterize bacterial population and its risk factor in raw milk.
The advent of high-throughput sequencing technology facilitates the inquiries in the field of micro-ecology that had been deterred due to technological limits. In this study, high-throughput sequencing technology was used to study the bacteria population structure and diversity in raw milking procedure and dairy farm environment, predicting the source of bacterial contamination in raw milk. The results of this study can be used to predict the possible bacteria species in raw milk, provides the basis for good hygienic practices and standardized operation procedures in the milk production to deliver high-quality milk products.
Material and Methods
The study was conducted in a large dairy farm (more than 1,500 cows in the stockade) in Tangshan City, Hebei Province, China (118.02°E, 39.63°N). The Milking Parlour, milk-storing hall and Sports Ground were selected as sampling sites. The sampling sites were cleaned and rinsed daily after milking, samples collected included pre-sterilized cow’s teats (C1), post-sterilized cow’s teats (C2), milking cluster (E), milk in storage tank (M1), milk in transport vehicle (M2), milk storage equipment (E2), cow dung samples (F) and cow’s drinking water (W). The samples were stored in liquid nitrogen tank for a short time after collection. Study was performed in September of 2019, and samples were taken 3 times per day, for a consecutive 5 days. The samples collected at the same day was mixed together and serve as a replicate. For each site, 5 repeats were sequenced and analyzed.
In this study, 8 typical sites in dairy farm in Hebei province of China were selected, the bacterial population composition and diversity were studied by high-throughput sequencing. Samples were collected in the experiment dairy farm. Collection of samples C1.1–C1.6: six healthy cows were randomly selected, samples were taken with sterile cotton swab from the area of 1 cm2 around the teats, and then placed in 10 mL sterile normal saline immediately; collection of samples C2.1–C2.6: Samples were taken with sterile cotton swab from the cows corresponds to C1 in the same way; samples collection of E.1–E.6: according to the distribution of the milking cluster, 6 milking cluster that can cover the whole milking parlor were selected, and the surface of the clusters were smeared with sterile cotton swabs for 3 times and placed in sterile saline solution immediately. Collection of M1.1–M1.6 and M2.1–M2.6 samples: after proper blending, the liquid milk bucket was used to collect milk from the surface, the middle and the bottom of the 3 points then thoroughly mixed and evenly, respectively 15 mL milk was taken and divided into 6 sample collection tubes; collection of E2.1–E2.6 samples: wiping with sterile cotton swab and placed in 10 mL sterile normal saline. Collection of F.1–F.6 samples: 6 different directions of cow’s sports field were selected, collect the excrement about 1g that does not have the impurity and put it into aseptic test tube with aseptic medicine spoon. Collection of W.1–W.6 samples: after each sampling site >10L breeding water was filtered, the filter membrane (cut or removed) was transferred to a sterile centrifuge tube for storage and inspection. All samples were stored in liquid N2 for long-term preservation immediately after collection.
DNA was extracted from 48 samples of 8 groups. The V3–V4 hypervariable region of 16S rRNA were amplified by PCR for barcoded pyrosequencing. The 16S rRNA gene V3–V4 region of bacteria was amplified using the universal Forward: 5’-ACTCCTACGGGAGGCAGCAGCAG-3’ and reverse 5’-GGACTACHVGGGTWTCTAAT-3’. The PCR amplification products were purified and dissolved in Elution Buffer by Agencourt AMPure XP magnetic beads, and then the library was constructed. Agilent 2100 Bioanalyzer was used to detect the range and concentration of fragments in the library, the V3–V4 region of the 16S rDNA gene was amplified with the qualified sample DNA as the template, and the magnetic beads were used to screen Amplicon fragments. Finally, the qualified library was used for Cluster preparation and Paired-end sequencing. The data obtained from the computer were used for the corresponding bioinformatics analysis (Avershina et al., 2013; Smith and Peay, 2014).
The data from Illumina platform was filtered to remove the low-quality sequences and the remaining high-quality valid sequences, which can be used for subsequent analysis (Fadrosh et al., 2014; Sørensen et al., 2016); FLASH V1.2.11 software was used to assemble the paired sequences obtained by paired-end sequencing into a sequence by overlapping relationship, and tag sequences with high variable region were obtained. The minimum matching length was set to 15 bp and the allowable mismatch rate of Overlap region was 10%. Sequences without overlapping relationship were removed (Cicconi-Hogan et al., 2013; Magoč and Salzberg, 2011); USEARCH V9.1 was used to cluster the splice effective sequences with 97% similarity, and then the OTU representative sequences were compared with the Greengene database by RDP Classifer V2.2 software, and the species annotation of OTU was carried out (Edgar, 2013; Edgar et al., 2011; Fouts et al., 2012; Wang et al., 2007); based on the results of OTU and species annotation, species complexity analysis and inter-group species difference analysis were performed.
The α-diversity of 8 groups was calculated using the VEGAN package in R 3.4.3, and the following indices were analyzed: observed species, Chao, Ace, Shannon, Simpson, and Coverage. The α-diversity values of the samples were calculated using the Mothur (v1.31.2) software, and the corresponding rarefaction curves, Heatmap analysis β-Diversity heatmaps and Clustering trees were generated using the R (v3.1.1) software. Principal component analysis (PCA) was conducted to compare similarities among samples using R and the corresponding rarefaction curves were generated using the R (V3.1.1). Intergroup differences in alpha-diversity indices were presented as box plots. Histograms were constructed for all taxa at the genus level. Cluster analysis was performed using the QIIME (v1.80) software. An iterative algorithm was used to perform sampling of 75% of the sequences in a sample with the least number of sequences using weighted and unweighted taxon abundance data, respectively. The final statistical results were obtained by analyzing the overall statistics after 100 iterations. The clustering method used was the unweighted pair group method with arithmetic mean (UPGMA). The significance of intergroup differences was analyzed using the R software rank-sum test.
Results
The total viable count of the 8 tested sites varied substantially. Due to the nature of samples, the total viable count is measured separately in F, which was presented in different unit (CFU/g) and showed extraordinarily high Total viable bacterial counts (TVC; Table 1). Overall, E2 and W demonstrated the lowest TVC, C2, and E present moderate amount of TVC. C1, M1, and M2 demonstrated similar TVC. We can see that the TVC increases significantly from E to M1, and slightly from M1 to M2, indicating that additional measures are desired for the storage and transportation of milk.
High-throughput sequencing of 16Sr RNA (V3–V4 region) of bacterial genome was carried out on 48 samples from 8 different sampling sites, and the composition of bacterial population was obtained. As shown in Fig. 1, the rarefaction curves of all samples had reached plateaus with the current sequencing, and the species had no more obvious increase as the sample number increased, which indicated that the sequencing depth and coverage was sufficient and the sample volume in our study was relatively large enough to reflect the species richness.
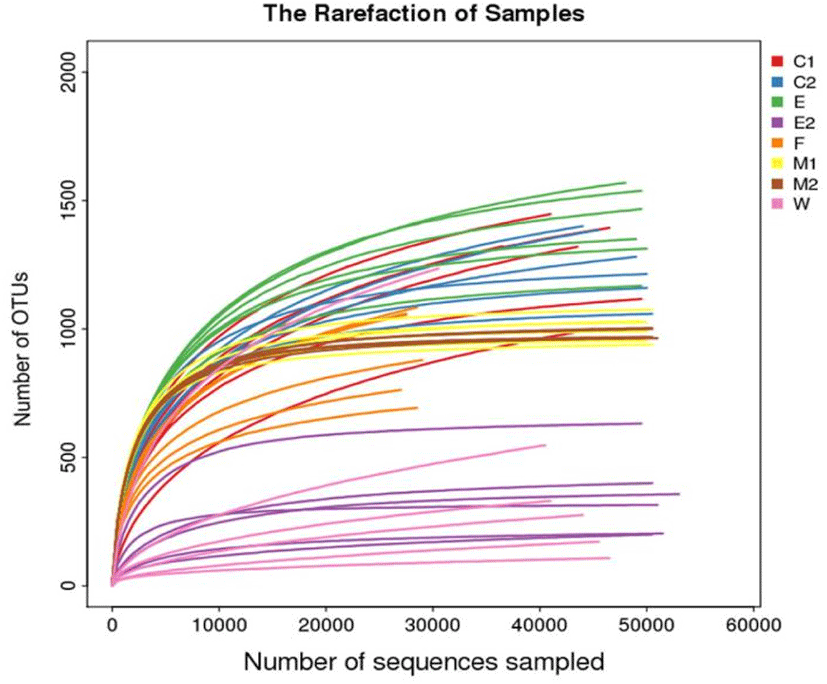
A total of 2,624,955 original sequences and 2,181,981 quality control sequences were obtained from the 48 samples at 8 different sampling sites. After clustering the merged tags, 45,726 OTUs were obtained from the 16S rRNA data at 97% similarity. Among them, the OTUs in E group were the most, reaching 8,408 OTUs, while the OTUs in E2 group were the least, only 2, 108 OTUs (Table 2).
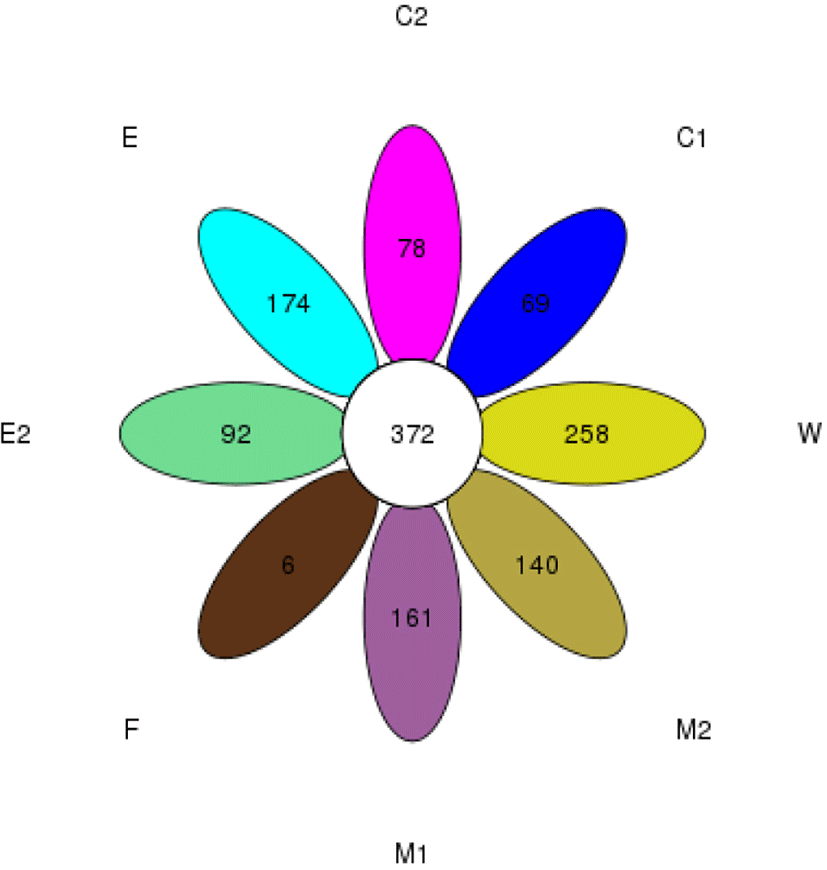
Among the 48 samples in 8 sampling sites, the common number of OTUs in 8 groups is 372, which accounted for 4.4%–17.6% of the total number of OTUs in each group, of which C1 group has 69 unique OUTs, C2 has 78 unique OTUs, E has 174 unique OUTs, M1 has 161 unique OTUs, M2 has 140 unique OTUs, E2 has 92 unique OTUs, F only has 6 unique OTUs and W has 69 unique OTUs, which accounting for 0.91%, 1.04%, 2.07%, 2.68%, 2.38%, 5.69%, 0.11%, 2.58% of the total OUTs, respectively (Fig. 2). In addition, the results also showed that among the 8 groups, E (milking equipment) group had the most unique OTUs, indicating that E group is most diverse in bacterial populations and post a key factor influencing the quality of milk.
The Alpha diversity indices of 8 groups were as shown in Table 3, and there were significant differences (p<0.05, respectively). The bacterial population richness of C1 and E group were the highest among all sampling sites, and Chao index, Ace index and Shannon index of C2, M1, M2, F groups were significantly higher than E2 and W groups, but Simpson index of C2, M1, M2, F groups was significantly lower than that of E2 and W groups. The Shannon index of E, M1 and M2 was higher than that of the other groups, which indicated that the bacterial population of the milking cluster and raw milk samples had higher diversity, and the species diversity of E2 and W was the lowest.
Comparison of OTUs against the database at the phylum, class, order, family, genus, and species levels resulted in the annotation of the 16S rRNA sequence-based OTUs to 36 phyla, 96 classes, 186 orders, 353 families, 766 genus and 896 species.
The NGS method was used for comparative analysis with Greengene database. Approximately 36 phyla and 799 genera were detected. The predominant phylum was Firmicutes which account for 32.36% (C1), 44.62% (C2), 44.71% (E), 41.10% (M1), 45.08% (M2), 8.08% (E2), 53.38% (F), 4.47% (W) in each group; proteobacteria was the subdominant phylum, which account for 20.72% (C1), 16.01% (C2), 17.39% (E), 15.49% (M1), 13.06% (M2), 21.88% (E2), 3.98% (F). Proteobacteria was the absolute dominant phylum accounting for 81.79% in W group; actinobacteria accounts for 56.43% in E2 group. Minor phyla in 8 groups including Bacteroidetes and Actinobacteria (Fig. 3A).
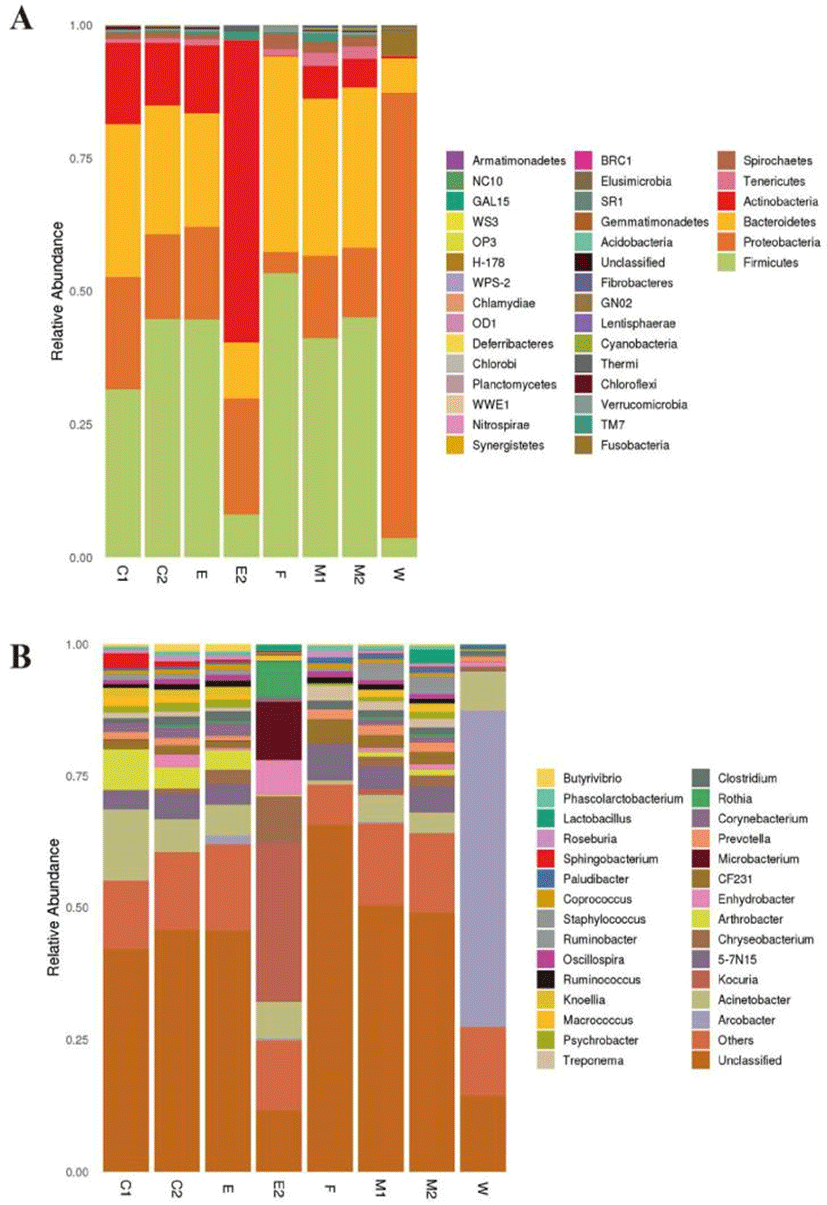
The dominant genus in 8 groups were Acinetobacter, Arthrobacter, Kocuria, Chryseobacterium, Clostridium, Corynebacterium, Enhydrobacter, Microbacterium, Prevotella, Macrococcus. Considerable difference was noted between the bacterial compositions of the 8 groups. The bacterial composition in C1, C2, and E is most similar, the most abundant genus were Acinetobacter, Arthrobacter and Sphingobacterium. The dominant bacterial genera were Kocuria, Microbacterium and Chryseobacterium in E2 group, which account for 30.04%, 10.89% and 8.69%, respectively; The predominant genera was Acinetobacter in 8 groups which accounted for 13.06% (C1), 6.31% (C2), 5.84% (E), 5.04% (M1), 3.90% (M2), 6.96% (E2), F (0.81%), W (7.56%) at each sampling sites. Arthrobacter was the subdominant bacteria genera, which accounted for 7.43% (C1), 4.01% (C2), 3.65% (E), 0.25% (E2), 0.86% (M1), 1.06% (M2), F (0.01%), W (0.02%) at each sampling site, Ranking the third dominant bacteria genera was Sphingobacterium, which accounted for 2.69% (C1), 1.03% (C2), 0.49% (E), 0.14% (E2), 0.17% (M1), 0.18% (M2), F (0.02%), W (0.03%) in each sampling site, respectively (Fig. 3B).
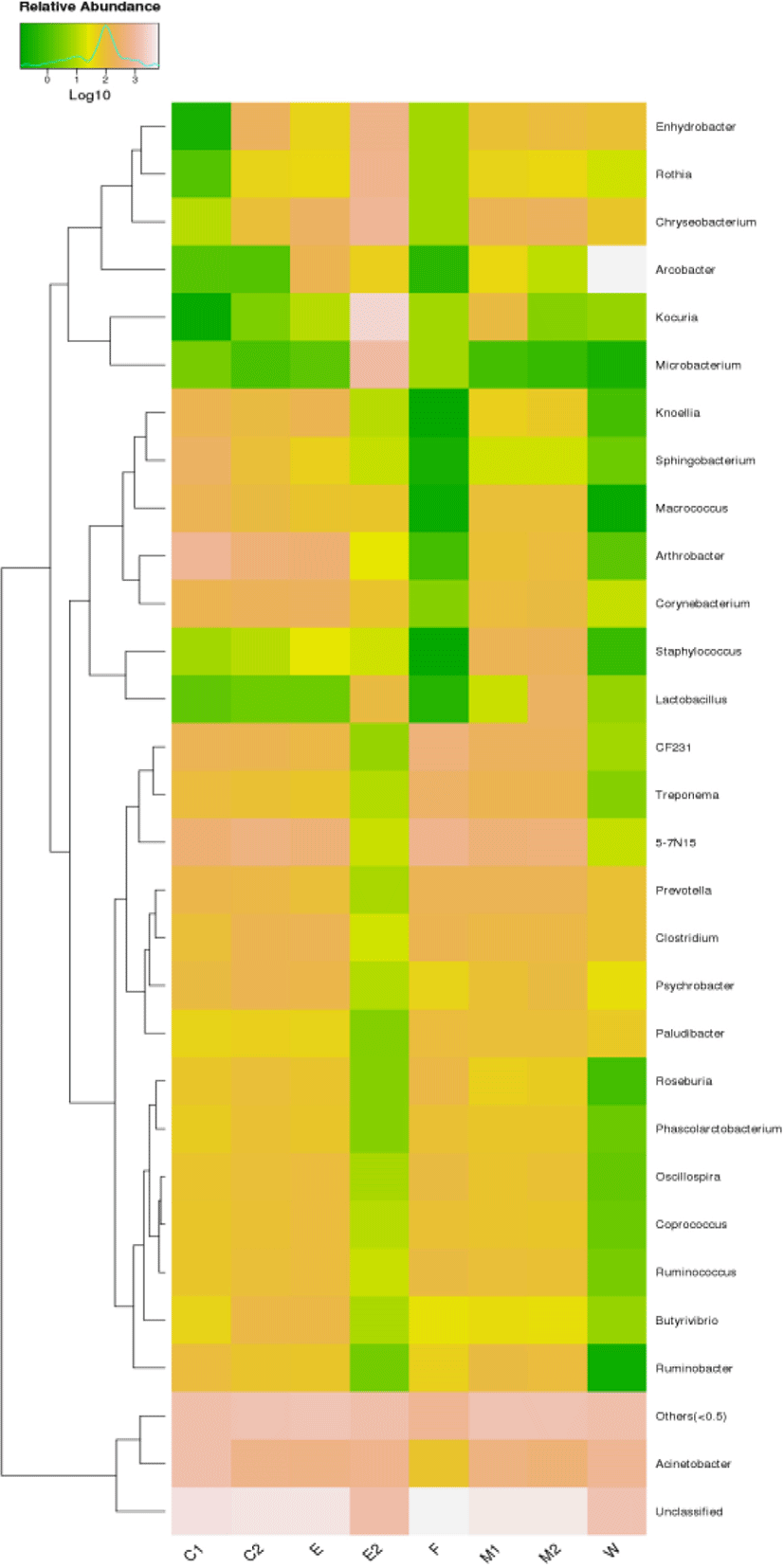
Heatmap clustering analysis were performed at the genus level, and all taxa with an abundance of less than 20% in a sample were group at others. The top 10 most abundant bacterial species, based on the 16S rRNA sequences, were in descending order of Acinetobacter, Arthrobacter, Sphingobacterium, Macrococcus, Corynebacterium, Knoellia, Psychrobacter, Ruminobacter, Kocuria, Chryseobacterium. The bacteria population of the collected samples was vertically clustered into two large branches according to the evolutionary relationship. Among the 8 group, C1, C2, and E were relatively close to each other in the graph, which shows that the diversity of species composition is small. The TOP3 bacteria population were Acinetobacter C1 (13.06%), C2 (6.31%), E (5.84%), Arthrobacter C1 (7.43%), C2 (4.01%), E(3.65%) and Corynebacterium C1 (1.57%), C2 (2.11%), E (2.36%). However, Chryseobacterium was the predominant genus in M1 and M2 group, which account for M1 (1.96%), M2 (2.26%), Staphylococcus was the subdominant genus in M1, M2 groups, which account for M1 (1.91%), M2 (2.15%). The top 3 dominant genus were Kocuria (30.04%), Microbacteria (10.89%) and Rossia (6.92%) in E2 group, while the predominant genus was Arcobacter (57.65%) in W group, which indicated that the bacterial population composition in group E2 and W was quite different from that in other groups (Fig. 4).
Cluster analysis showed that the bacterial population compositions of the M1 and M2 were quite similar, the bacterial population compositions of the C1, C2, and E were quite similar, but E2 group and W group differs in species composition from the other 6 groups (Fig. 5).
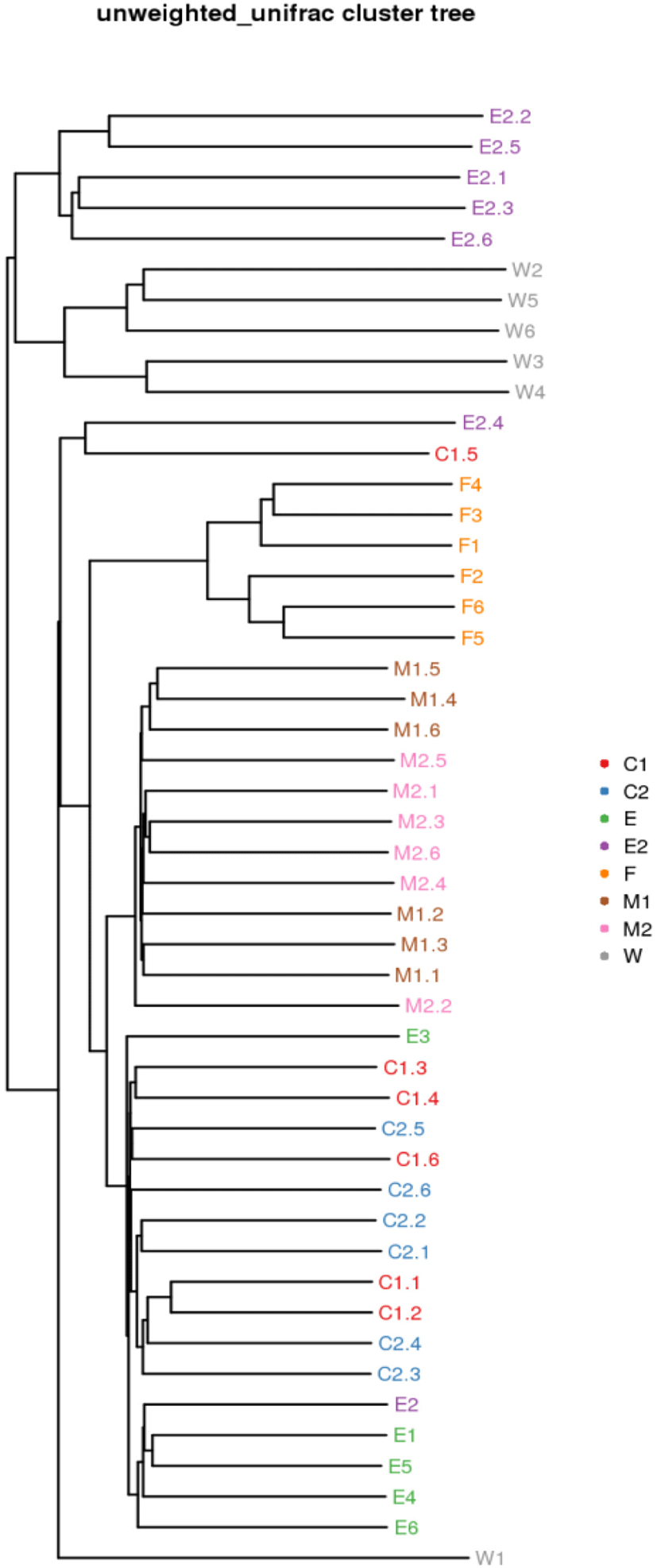
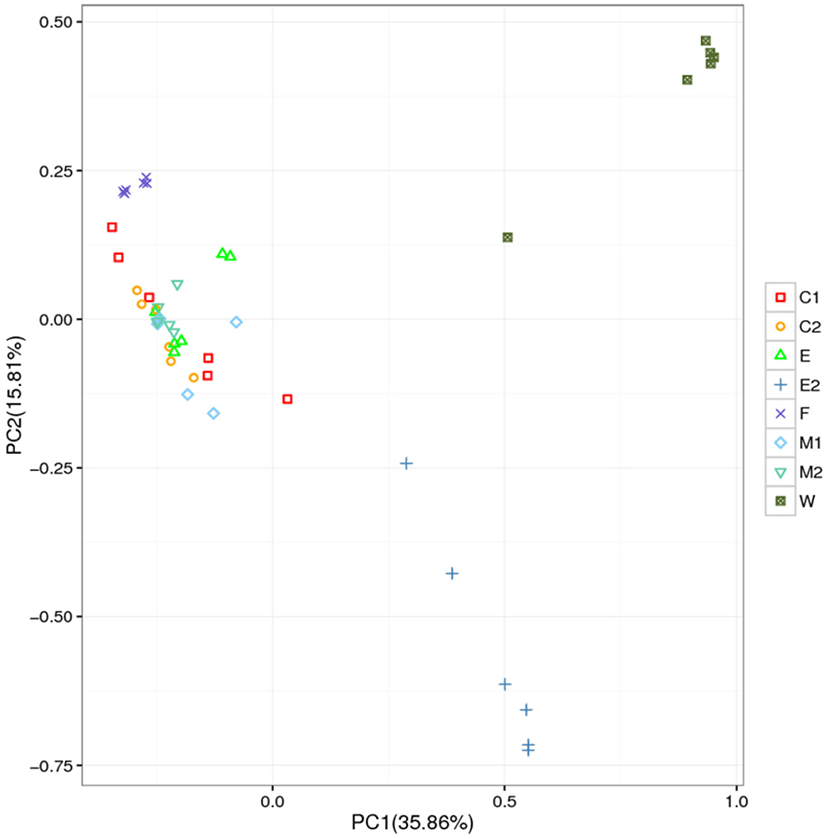
PCA was performed based on the OTUs abundance. The composition of bacteria population in two raw milk (M1 and M2 group) were very close in the figure, and some sites almost overlapped. In addition, the bacterial population in C1, C2, and E were relatively similar. However, there were significant differences between the W, E2 and other 6 groups in the bacterial population compositions (Fig. 6). The bacterial population structure of the 8 groups showed an obvious clustering phenomenon, with most of them clustered to the left and only a few to the right.
Discussion
High-throughput next-generation sequencing, also known as “next generation” or “deep” sequencing, which can sequence hundreds of thousands to millions of DNA sequences in one time, so it is also called deep sequencing (Ercolini et al., 2012; Quigley et al., 2012). In recent years, high-throughput sequencing technology has been widely used in the study of dairy products, gradually changing from the identification of dominant flora to the studies on the overall diversity of microorganisms (Abriouel et al., 2008; Liu et al., 2015). Due to the complexity of the dairy chain, microbial contamination can occur in different steps of production, leading to the development of adequate control plans for monitoring the microbial quality and safety of milk since production to processing (Wouters et al., 2002). Through high throughput sequencing technology, the key nodes of whole milking procedure which affect raw milk quality were deduced, and the key influencing factors of raw milk quality in the feeding environment of dairy farms were clarified.
Milk in healthy udder cells is thought to be sterile (Johnson et al., 2015), but there after becomes colonised by microorganisms from a variety of sources, including the teat apex, milking equipment, air, water, feed, grass, soil and other environments (Vacheyrou et al., 2011). Previous study found several microbial groups in different milking sites, some groups were used to assess the hygienic procedures and conditions during milking, such as Mesophilic aerobes and Coliforms (Wouters et al., 2002), some groups were considered as relevant spoilage agents, such as Sphingobacterium, Pseudomonas, and Clostridium; many bacteria were researched due to their pathogenic potential, such as Acinetobacter, Arthrobacter, Staphylococcus, Campylobacter and Arcobacter, and other bacteria can possess beneficial features, like some Lactobacillus, Lactococcus, Streptococcus and Enterococcus (Vacheyrou et al., 2011). This huge diversity is a challenge in the dairy industry, addresses their sources in different production procedure, which can guide the raw milk utilization by consumers and dairy industry.
This study presented a novel investigation of the bacterial population in china dairy farms. The predominant phylum was Firmicutes which account for 32.36% (C1), 44.62% (C2), 44.71% (E), 41.10% (M1), 45.08% (M2), 8.08% (E2), 53.38% (F), 4.47% (W) in each group, The predominant genera was Acinetobacter in 8 groups which accounted for 13.06% (C1), 6.31% (C2), 5.84% (E), 5.04% (M1), 3.90% (M2), 6.96% (E2), F (0.81%), W (7.56%) at each sampling sites. Milking parlors as the very heart of every dairy and where milking process are concerned, hygiene is key (Wouters et al., 2002). Udder health is an essential component of quality milk, mastitis is the common disease found in dairy herds in China. Cow teats surface can contain a high diversity of bacteria, this study revealed that Acinetobacter (13.06%) and Arthrobacter (7.43%) were detected in C1 but Acinetobacter (6.31%) and Arthrobacter (4.02%) in C2, there is a significant decrease in bacterial richness. Notably, teats disinfection is very important before milking which can reduce the diversity and richness of bacteria population. Previous study also shown that the use of some disinfectant products for pre-milking teat dip preparation can have beneficial effects on reducing the levels of staphylococcal and streptococcal pathogens on teat skin (Gleeson et al., 2009). Jones and Newburn (2002) found the two basic principles of mastitis control are first, elimination of existing infections and, secondly, prevention of new infections. Milking cluster (E) which can direct touch cow’s udder, incomplete cleaning can lead to a risk of mastitis, so, its cleanliness can directly affect the quality of raw milk. According to the results of Samples Clustering (Description, Weighted_Unifrac) and PCA, there was a notable clustering phenomenon toward the C1, C2, and E which may have been caused by the bacterial population composition of the C1, C2, and E were quite similar, it revealed that much of variance in bacterial communities of above 3 groups was associated with cleanness of cow teats and cleanness of milking cluster. The Top 3 dominant bacterial genus in F group were Treponema (2.84%), Prevotella (1.97%), Clostridium (1.60%), this study shows composition of F group was similar to the C1, C2, E, M1, and M2 groups, which further indicated that faeces could not be cleaned in time, microorganisms can cross-contaminate the milking cluster by adhering to the cow’s body or by air flow. The top 3 dominant genus in E2 group were Kocuria (30.04%), Microbacteria (10.89%) and Rossia (6.92%), while the predominant genus was Arcobacter (57.65%) in W group, which indicated that the bacterial population composition in group E2 and W was quite different from that in other groups. Our study showed that Acinetobacter (5.04%), Chryseobacterium (1.96%) and Treponema (1.68%) were the dominant genus in M1. However, Acinetobacter (3.90%), Lactobacillus (2.62%), Chryseobacterium (2.26%) were the dominant genus in M2. Cluster analysis showed that the bacterial population composition of M1 and M2 were quite similar, the results are partly consistent with previous studies, Lafarge and Hagi believed that there were two main strains in the milk, Lactobacillus and Staphylococcus (Hagi et al., 2010; Hagi et al., 2013). Delbès et al. (2007) detected that the dominant bacteria in milk were Clostridium and Lactobacillus. Previous study shows that the dominant bacteria detected in the commercial milk were Acinetobacter and Pseudomonas. In addition, cold-resistant bacteria are the main spoilage bacteria in milk, and Gammaproteobacteria and BacillusI are also the dominant bacteria with contents more than 1% in this study (Raats et al., 2011). Rasolofo et al. (2010) believed that the abundance of these two bacteria would increase with the prolongation of refrigeration time, so the processing time of raw milk into commercially available milk should be shortened. Milk storage equipment (E2) can contain a reservoir of bacteria, this study detected that Kocuria (30.04%), Chryseobacterium (8.69%) and Enhydrobacter (6.64%) were the dominant genus bacteria in E2. The bacterial population composition of E2 differs from other 7 groups, the reason for this difference may be caused by the tempe rature of the milk storage equipment and the microorganisms in the environment.
In conclusion, the difference of bacteria species diversity in different sampling sites may be related to the environmental health status of each space, the timely cleaning and wiping of bovine body, the sterilization of milking cluster and the transmission of aerosol pollution. In this study, a variety of bacteria genera were identified, including some pathogenic bacteria genera such as Acinetobacter, Arthrobacter, Sphingosinolium, Staphylococcus, Pseudomonas and Corynebacterium which were the main dominant bacteria genus in different milking sites. Acinetobacter and Corynebacterium can cause bovine mastitis, Sphingomonas can decompose milk fat and milk protein and remove low milk protein activity. Pseudomonas aeruginosa can also cause mastitis in cows. Bacillus anthracis can produce enterotoxin, which is highly pathogenic to humans and animals (Ercolini et al., 2012; Quigley et al., 2012). Acinetobacter as a kind of conditional pathogenic bacteria causing the cow’s mastitis, among 8 groups C1 (the cow teats before disinfection) with the highest percentage (13.06%), followed by W (7.56%), E2 (6.96%) and C2 (6.31%), the result suggests teats disinfection before milking is crucial and the cleanliness of the milk storage equipment also affects the quality of raw milk, the results also indicated that the cleanliness of drinking water in the farm directly affected the quality of raw milk.
In summary, in the traditional dairy farms of China, there are two factors can affect the quality of raw milk, one is the milking procedure, the other is the environmental sanitation. Milking procedure includes cow’s teats, milking cluster, milk storage equipment, milk from milk storage tanks and milk from transportation vehicles, the sampling sites in this study were C1, C2, E, E2, M1, and M2, while the farm environment mainly includes faeces and water, sampling sites were F and W in this study, based on the results of our study, bacterial population composition in different sampling sites of milking was significantly different, therefore, we believe that there is a considerable correlation between the proper milking procedure and raw milk quality. The timely disposal of excrement and the cleanliness of drinking water also helpful to guarantee the quality of raw milk, affect the quality of raw milk, pathogenic bacteria of messy environment in the dairy farms will through the injured cow nipple cause mastitis, therefore, it is necessary for the quality of raw milk to be ensured by the proper milking and the hygienic condition in the course of dairy cow breeding.
China has formulated and implemented a nationwide raw milk quality and safety testing plan since 2008, but compared with the development needs of dairy industry, the systematic research is still weak. This study from the perspective of industrial chain, systematically analyzed the effect of milking behavior and environment on the quality of raw milk in diary farm. About 90% of the bacterial communities which cannot be isolated in lab were obtained through high-throughput sequencing, this study gave a comprehensive and in-depth understanding of the bacterial diversity and composition along milking in dairy farms. It is of great significance to grasp the key nodes in the milk production process as a whole and provide a strong scientific basis for the quality and safety supervision of raw milk.