










Introduction
Meat is vulnerable to degradation from oxidation and microbial contamination, given its nutrient richness and favorable conditions for microorganisms (Lee and Yoon, 2024; Yu et al., 2018). Meat preservation is associated with food safety and meat spoilage. Thus, various research aimed to address the microorganisms responsible for these issues (Devlieghere et al., 2004). Meat spoilage occurs because of various microorganisms, depending on storage temperature, contamination, and packaging conditions. Enzymatic activity in animal muscle cells is a natural occurrence in animal muscle cells after slaughtering and contributes to meat spoilage (Dave and Ghaly, 2011). Key enzymes involved in this process include NADPH oxidase, cyclooxygenase, xanthine oxidase, nitric oxidase, and peroxidases (Bekhit et al., 2021b). Despite the implementation of cold chain systems to mitigate these concerns, vulnerabilities persist in the meat cold chain (Kwon et al., 2022); also, undesirable changes (e.g., protein decomposition, lipid oxidation, discoloration, and the growth of spoilage bacteria) may occur as meat spoilage progresses.
Volatile basic nitrogen (VBN) can be used as an indicator of meat spoilage and to assess the freshness of fish (Huang et al., 2015; Jeong et al., 2015). Thus, the VBN could be a critical consideration at the purchase stage. These compounds are produced through the microbial degradation of protein and non-protein nitrogenous substances, such as amino acids and nucleotide catabolites (Liu et al., 2013).
With recent advancements in gene analysis technology for identifying and characterizing microorganisms, it is now possible to predict meat quality based on microbiota (Gagaoua et al., 2022). Since meat spoilage is related to microorganisms in the meat, researches have been conducted to identify potential microorganisms for VBN production (Fang et al., 2022; Saenz-García et al., 2020; Wang et al., 2017).
Therefore, the objectives of this study were to investigate the prevalence of microorganisms and VBN in beef and to suggest potential bacteria that might contribute to VBN production in current purchase stages with metagenomic analysis.
Materials and Methods
Seventy beef samples [34 (high grade: 17 and low grade: 17) sirloin (relatively higher fat content) and 36 (high grade: 19 and low grade: 17) top round (relatively lower fat content) samples] were collected from the wholesale stage of distribution (17%), butcher’s shops (41%), hypermarkets (17%), and supermarkets (24%) between July and August 2022. These distribution stages and portions for each retail outlet were determined according to the data from the Korea Institute for Animal Product Quality Evaluation (KAPE, 2022), and the distribution stages were also where the consumer purchased beef in current distribution conditions. All collected beef samples were transported in a cooler and analyzed within 3 h of purchase.
According to the analysis method by the Ministry of Food and Drug Safety (MFDS, 2022), for qualitative analysis of Escherichia coli, 25 g of beef samples were aseptically placed in a filter bag containing 225 mL sterile 0.1% buffered peptone water (BPW; Becton, Dickinson, and Company, Detroit, MI, USA), and homogenized with a pummeler (BagMixer®, Interscience, St. Nom, France) for 1 min. The homogenate was diluted with 0.1% BPW, and 1 mL aliquots of the diluents were placed into 9 mL EC medium (Becton, Dickinson, and Company) and incubated at 44°C for 24 h. A positive sample was identified by turbidity and gas production in the EC medium. Subsequently, a loopful of the positive EC medium was streaked on eosin methylene blue (EMB; Becton, Dickinson, and Company) agar and incubated at 37°C for 24 h. Colonies displaying a green metallic sheen were subjected to identification through 16S rRNA sequencing. To confirm colony formation of enterohemorrhagic E. coli (EHEC) with a method by the MFDS (2022) with the modification, 10 g of the beef sample was aseptically placed in a filter bag containing 90 mL modified tryptic soy broth (mTSB; MBCell, Kisanbio, Seoul, Korea). The sample was placed at 37°C for 24 h for the enrichment of EHEC. A loopful of the culture was streaked onto MacConkey sorbitol agar (Becton, Dickinson, and Company) supplemented with cefixime tellurite (MBCell; TC-SMAC), and 5-Bromo-4-Chloro-3-Indolyl-β-D-Glucuronide (BCIG) agar (Oxoid, Basingstoke, UK). The agar plates were incubated at 37°C for 24 h. After the colony formation of EHEC was confirmed, the following experiment was conducted to detect the DNA of EHEC. To extract DNA from the colonies on the agar plates after incubation, a method by Fratamico et al. (2000) was used with the modification. Two to four red colonies on TC-SMAC and turquoise colonies on the BCIG agar were each suspended in 100 μL of sterile distilled water, incubated at 99°C for 10 min, and the resulting mixture served as the template DNA for polymerase chain reaction (PCR) amplification with multiplex PCR. The PowerchekTM diarrheal E. coli 8-plex detection kit (Kogene Biotech, Seoul, Korea) was used to detect stx1 and stx2, which are specific DNA markers for EHEC. PCR amplification was conducted according to the manufacturer’s instruction and consisted of initial denaturation at 95°C for 12 min, followed by 32 cycles of 95°C for 30 s, 60°C for 45 s, 72°C for 60 s, and a final extension at 72°C for 10 min. The PCR products were electrophoresed on 2% agarose gel, and DNA bands were visualized under UV light.
The homogenates previously prepared for qualitative analysis of E. coli were diluted in 9 mL of 0.1% BPW and used for quantitative analysis of microorganisms. The diluents were plated on PetrifilmTME. coli/Coliform Count Plates (3M, Saint Paul, MN, USA), Palcam agar (Oxoid), xylose lysine deoxycholate agar (XLD; Becton, Dickinson, and Company), Baird-Parker agar (BPA; MBcell) supplemented with egg yolk tellurite (MBcell), PetrifilmTM Aerobic Count plates (3M), PetrifilmTM Enterobacteriaceae Count Plates (3M), de Man, Rogosa, and Sharpe agar (MRS agar; Becton, Dickinson, and Company), cetrimide agar (Becton, Dickinson, and Company), plate count agar (PCA; Becton, Dickinson, and Company), and PetrifilmTM Yeast & Mold Count Plates (3M) for E. coli and coliform, Listeria monocytogenes, Salmonella, Staphylococcus aureus, total aerobic counts, Enterobacteriaceae, lactic acid bacteria, Pseudomonas spp., psychrotrophic bacteria, and yeast and mold, respectively. PetrifilmTME. coli/Coliform Count Plates, BPA supplemented with egg yolk tellurite, and PetrifilmTM Aerobic Count plates were incubated at 37°C for 48 h. Palcam agar and cetrimide agar were incubated at 30°C for 48 h. XLD and MRS agar were incubated at 37°C for 24 h. PetrifilmTM Enterobacteriaceae Count Plates were incubated at 37°C for 48 h. PCA and PetrifilmTM Yeast & Mold Count Plates were incubated at 7°C for 10 d and at 25°C for 5 d, respectively. The cell counts of the following bacteria were determined based on the colonies identified through the following methods and 16S rRNA sequencing. For the identification of E. coli, blue colonies that produced gas on PetrifilmTME. coli/Coliform Count Plates were streaked onto EMB agar. Colonies exhibiting a green metallic sheen after 24 h of incubation at 37°C were isolated. For L. monocytogenes, colonies presumptively identified on Palcam agar plates were streaked to CHROMagarTM Listeria (CHROMagar, Paris, France) and incubated at 37°C for 24 h. Colonies appearing blue with a diameter of less than 3 mm and displaying a regular white halo on CHROMagarTM Listeria were isolated. Since no colonies indicative of Salmonella were observed, no further analysis was conducted for this bacterium. For S. aureus, colonies presumptively identified on BPA supplemented with egg yolk tellurite were streaked onto CHROMagarTM Staph aureus (CHROMagar) and incubated at 37°C for 24 h. Pink to mauve colonies were isolated. For Pseudomonas spp., colonies on cetrimide agar were streaked onto CHROMagarTM Pseudomonas (CHROMagar) and incubated at 30°C for 24–36 h. Blue-green colonies were isolated. All the isolated colonies were then subjected to the 16S rRNA sequencing. The 16S rRNA sequencing was performed by BIONICS (Seoul, Korea) using universal primers 27F and 1492R. The resulting sequences were analyzed by comparing them with microbial sequences in the NCBI GenBank database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) using the BLAST (Basic Local Alignment Search Tool) to identify the bacteria in BIONICS. In this experiment, the presumptive colonies on the media where the homogenates were plated were streak-plated on the second media with higher selectivity, and isolated colonies on the second media were identified by 16S rRNA sequencing. Based on this identification, only identified colonies of the presumptive colonies were counted.
VBN contents were also analyzed for samples corresponding to microbiological analysis. VBN content was evaluated with the micro-diffusion method (Conway and O’Malley, 1942; MFDS, 2022). In brief, a sample bag containing 5 g beef and 25 mL distilled water was homogenized with a pummeler for 1 min, and the homogenate was left at room temperature for 30 min. The homogenate was then filtered using Qualitative Filter Papers No. 131 (Advantec, Tokyo, Japan). Subsequently, 1 mL of 0.01 N sulfuric acid (H2SO4; Daejung Chemicals & Metals, Siheung, Korea) was placed into the inner chamber of the Conway diffusion cell (Daihan Scientific, Wonju, Korea), and 1 mL of the filtrate and 1 mL of a saturated K2CO3 solution (Samchun Chemical, Seoul, Korea) were placed into the outer chamber. The Conway diffusion cell was sealed with glycerin and incubated at 25°C for 1 h. After incubation, the VBN-captured H2SO4 solution was titrated with 0.01 N sodium hydroxide (Daejung Chemicals & Metals, Siheung, Korea) with the addition of 10 μL indicator solution to the inner chamber. To prepare the indicator solution, 0.1 g of methyl red (Duksan Pure Chemicals, Ansan, Korea) and 0.1 g of methylene blue (Sigma-Aldrich, St. Louis, MO, USA) were each dissolved in 100 mL of ethanol, filtered, and mixed in a 2:1 ratio (v/v). The following equation was used to calculate the concentration of VBN (MFDS, 2022).
W=sample weight, a=blank, b=sample, f=factor of 0.01N NaOH, DW=distilled water volume.
To analyze microbiota in beef corresponding to changes in VBN content, of 70 samples, three samples with the highest VBN content (VBNH) and three samples with the lowest VBN content (VBNL) were selected. To extract DNA from the sample, a method by Li et al. (2020) was used with some modifications. Each sample (25 g) was placed in a sample bag containing 225 mL sterile 0.1% BPW and pummeled for 1 min. Ten milliliters of the homogenate were spun down for 10 min, and the supernatant was transferred to a conical tube. The supernatant was then centrifuged at 5,000×g for 15 min at 4°C, and the pellet was resuspended with 10 mL of phosphate-buffered saline (PBS; pH 7.4; 0.2 g KH2PO4, 1.5 g Na2HPO4, 8.0 g NaCl, 0.2 g KCl/1 L distilled water). The suspension was centrifuged at 5,000×g at 4°C for 15 min. The pellet was then used for genomic DNA extraction. Genomic DNA was extracted from the pellet according to the manufacturer’s instructions using a DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany). For sequence library preparation, the Illumina 16S Metagenomic sequence libraries were prepared according to the Illumina 16S Metagenomic Sequencing Library protocols, and the V3 and V4 regions were amplified. Library preparation and paired-end sequencing were performed at Macrogen (Seoul, Korea) with the MiSeqTM platform (Illumina, San Diego, CA, USA). Sequencing results in FASTQ files were subsequently processed and analyzed with the 16S based microbiome taxonomic profiling pipeline of the EzBioCloud (CJ Bioscience, Seoul, Korea) for microbial community and diversity analysis. The PKSSU 4.0 version of the EzBioCloud was used as the reference database for the classification and identification of bacteria with a cut-off percentage set at 0.5% to exclude low-abundance taxa. Microorganisms identified below this threshold were classified into the et cetera (ETC) group. The cut-off was used to avoid the complexity in data analysis caused by including very low-abundance microorganisms, which may often result from sampling errors or other technical variances (Brumfield et al., 2020; Sadurski et al., 2024). Metagenomic analysis for yeast and mold was not conducted because they were analyzed with 16S rRNA, and thus, it was not appropriate to compare the relative abundance with bacteria. Additionally, yeast and mold populations were much lower than those of bacteria in beef samples.
Results and Discussion
Qualitative analysis showed no presence of E. coli and EHEC in beef (data not shown). Quantitative analysis also showed that E. coli, Salmonella spp., and L. monocytogenes counts were below the detection limit (<1.0 Log CFU/g; Table 1). Coliform counts were 1.7–2.1 Log CFU/g. S. aureus was detected at 1.2 Log CFU/g in only one out of 70 samples. Regardless of part, distribution channel, and grade, total aerobic bacteria levels were observed between 4.5 and 5.7 Log CFU/g, while Enterobacteriaceae ranged from 2.0 to 3.3 Log CFU/g. Lactic acid bacteria counts varied from 3.8 to 4.4 Log CFU/g. Pseudomonas spp. counts were in a range of 1.7 to 2.4 Log CFU/g. Psychrotrophic bacterial counts ranged from 4.7 to 6.2 Log CFU/g. Yeast and mold counts were from 2.9 to 3.3 Log CFU/g.
The cold chain system is used to delay meat spoilage by maintaining low temperatures during various stages, including post-slaughter carcass storage, cut handling, meat transport to distributors, and storage at retail sites (Ercolini et al., 2009). These practices might be related to the higher levels of psychrotrophic bacteria than those of other microorganisms in meat, as these bacteria could proliferate at refrigeration temperature. In contrast, the growth of microorganisms with higher optimal growth temperatures was inhibited under these conditions (Anas et al., 2019). This reason might cause somewhat higher psychrotrophic bacterial cell counts than the other bacteria. Some lactic acid bacteria in meat are psychrotrophic (Ercolini et al., 2009; Yost and Nattress, 2002), and Pseudomonas spp. are also psychrotrophic (Gill and Newton, 1978; Kim et al., 2013; Ledenbach and Marshall, 2009). Thus, their cell counts might contribute to relatively higher cell counts of psychrotrophic bacteria and total aerobic bacteria.
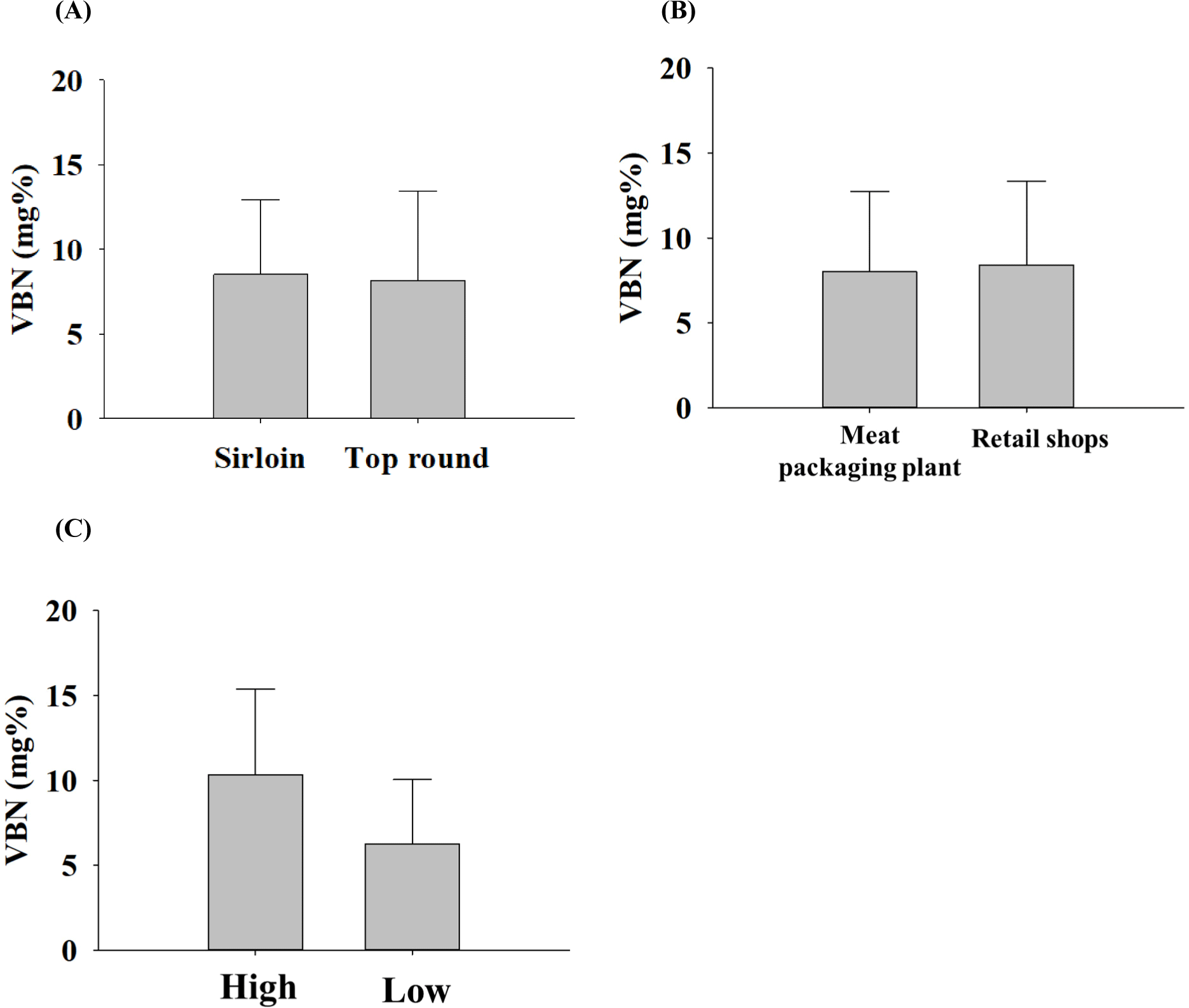
VBN serves as an indicator of protein or amine degradation, and its value was assessed to measure the freshness of beef (Bekhit et al., 2021a). On average, sirloin and top rounds exhibited 8.51±4.44 mg% and 8.15±5.33 mg% of VBN values, respectively (Fig. 1A). In distribution, the VBN values were 8.01±4.75 mg% and 8.39±4.95 mg% for meat packaging plants and retail shops, respectively (Fig. 1B). High-grade samples showed 10.33±5.04 mg% of VBN values, and low-grade samples had 6.26±3.79 mg% (Fig. 1C). In summary, VBN values did not depend on parts, distribution channels, and grades. However, it may vary depending on the sampling season, the number of samples, and the geographic region of sampling. In the other study, raw beef had a VBN content of 8.70±0.40 mg% (An et al., 2020), which is similar to the findings in our study. Based on this result, three VBNH samples and three VBNL samples were selected from 70 samples; their VBN values were 1.06±0.64 mg% and 20.73±2.79 mg% for VBNL and VBNH samples, respectively.
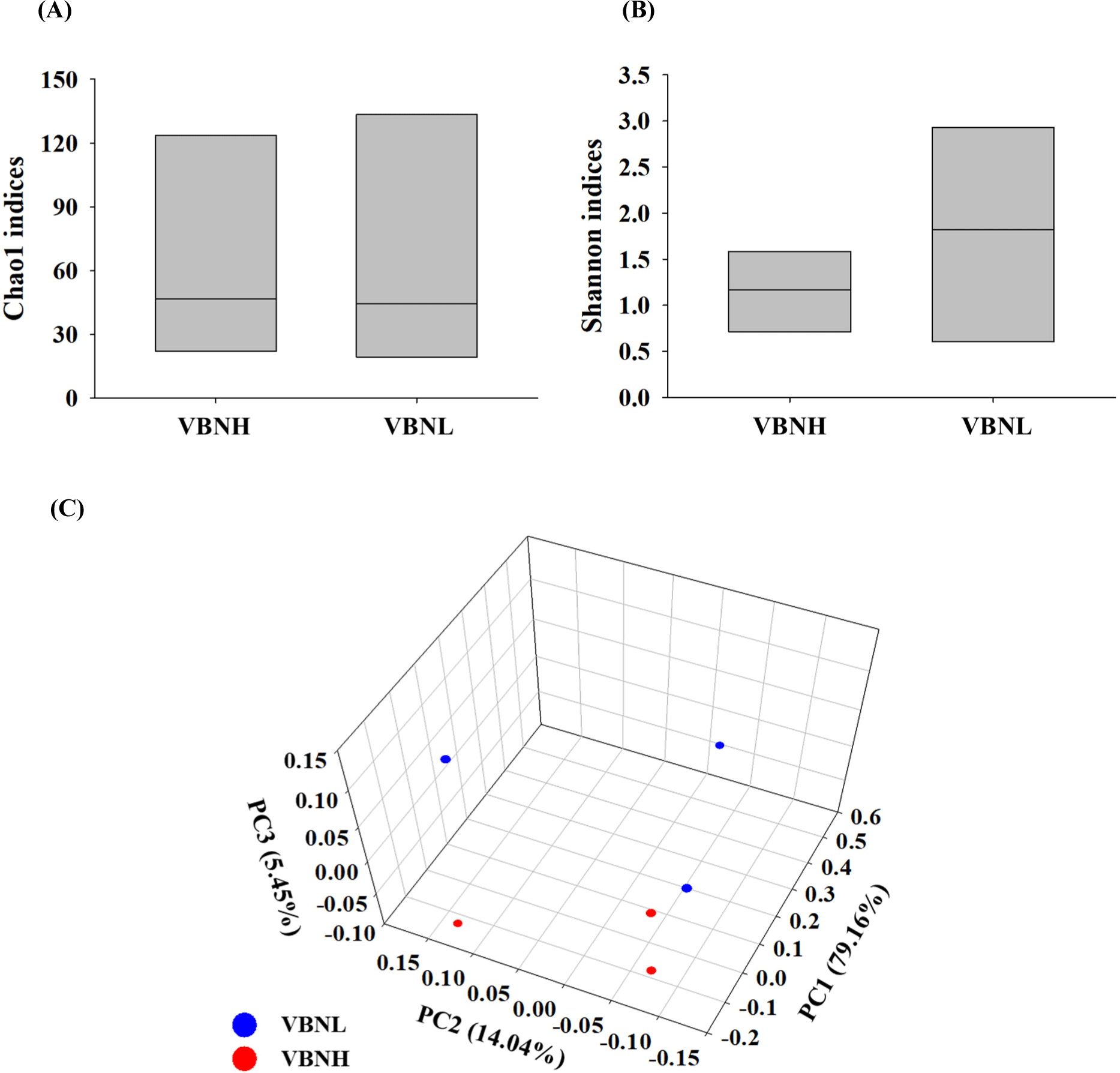
To compare the differences in beef microbiota by alpha diversity, the Chao1 (species richness) and Shannon (species diversity) indices were calculated (Chao et al., 2014; Figs. 2A and B). The Chao1 index did not reveal a significant difference between the VBNH and VBNL groups. However, the Shannon index was higher in the VBNL group than in the VBNH group, indicating higher species diversity in these samples. Beta diversity (the variation in species composition) using the principal coordinate analysis (PCoA) was conducted to compare microbial distribution (Legendre et al., 2005; Fig. 2C). Different microbial community cluster patterns were observed between the VBNH and the VBNL groups. The PCoA analysis showed that more spread was observed in the VBNL group, and it indicates a higher diversity in microbial composition. Specifically, the VBNL samples displayed a wider spread along the principal coordinate (PC), particularly PC1 and PC2.
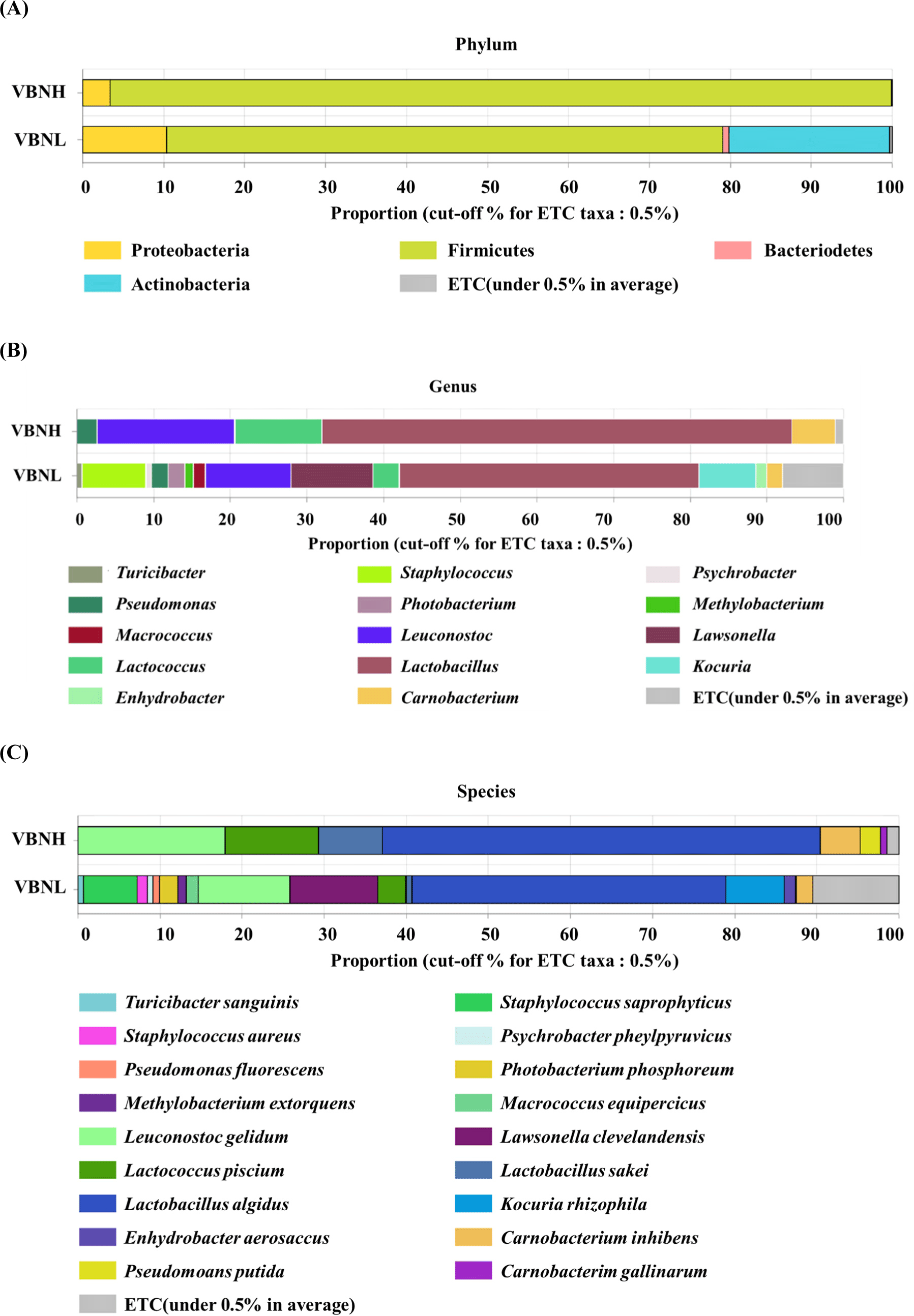
At the phylum level, Firmicutes and Proteobacteria were identified as common phyla in all samples (Fig. 3A). The VBNH group comprised 96.6% Firmicutes, a minor presence of Proteobacteria, and other phyla of the total composition. Conversely, the VBNL group showed a more diverse microbiota composition: Firmicutes accounted for approximately 68.7%, Actinobacteria accounted for about 19.8%, and smaller proportions of Proteobacteria and other phyla. At the genus level, microbiota was also more varied in the VBNL group than in the VBNH group, and the VBNH group had relative abundance in order of Lactobacillus (61.3%)>Leuconostoc (17.9%)>Lactococcus (11.4%)>Carnobacterium (5.7%)>Pseudomonas (2.6%), and their relative abundances were higher compared to the VBNL group (Fig. 3B). These genera, Lactobacillus, Leuconostoc, Lactococcus, and Carnobacterium, are all lactic acid bacteria (Ringø and Gatesoupe, 1998). Lactic acid bacteria belong to the family of Firmicutes (Liu et al., 2010). Hence, this result may correspond to the comparison for Firmicutes and Proteobacteria, as the VBNH group showed a high relative abundance of Firmicutes with lactic acid bacteria. Among the Lactobacillus species, which were found in high proportions in both VBNH (61.3%) and VBNL (39.1%) groups in our study, most of them frequently isolated from meat and meat products are psychrotrophic bacteria (Ercolini et al., 2009; Morishita and Shiromizu, 1986). Similarly, certain Leuconostoc species, such as Leuconostoc gelidum and Leuconostoc gasicomitatum were also psychrotrophic bacteria frequently isolated from meat (Comi et al., 2024; Johansson et al., 2022; Mun et al., 2021; Shaw and Harding, 1984). At the species level, microbiota was more varied in the VBNL group than in the VBNH group (Fig. 3C). Lactobacillus spp. in the VBNH group were predominantly represented by Lactobacillus algidus and Lactobacillus sakei (Fig. 3C). The other notable species were Leu. gelidum and Lactococcus piscium. L. algidus is known as Dellaglioa algida (Poirier et al., 2018; Sun et al., 2015; Zheng et al., 2020), and it grows within a temperature range of 0°C–25°C (Kato et al., 2000). L. algidus has been isolated from various meat and dairy products, including cattle milk, cured seasoned pork, cured ripened sausages, cooked cured or seasoned pork, and bovine meat (Parente et al., 2023; Pothakos et al., 2014; Sakala et al., 2002; Stoops et al., 2015). L. sakei is also a psychrotrophic lactic acid bacteria commonly found in fresh meat and fish (Chaillou et al., 2005). Vihavainen and Björkroth (2007) identified Leu. gelidum and L. sakei as predominant populations in the lactic acid bacteria in packaged spoiled beef. This result from our study shows that microbiota differs between the samples with high and low VBN, especially for lactic acid bacteria such as Lactobacillus and Leuconostoc which are the first and second most. These bacteria are known to have proteolytic activity (García-Cano et al., 2019; Kieliszek et al., 2021), which may lead to protein degradation and the production of volatile nitrogenous compounds (Bekhit et al., 2021b; Thorn and Greenman, 2012). Thus, they might be related to producing more VBN contents in the VBNH samples in this study. However, this is just a potential suggestion because this result is based only on metagenomic analysis. The results of the metagenomic analysis show the relative plentifulness of certain bacteria rather than indicating an absolute relation of the higher relative abundance to the higher VBN production. Meat contains calpain, cathepsin, and caspases, which function as proteases, and alanine aminopeptidase, arginine aminopeptidase, and serine aminopeptidases which function as peptidases (Sentandreu et al., 2002; Toldrá and Flores, 2000). Decarboxylases in meat may be involved in the decarboxylation of amino acids, leading to the formation of VBN (Halász et al., 1994; Tosukhowong et al., 2011). These enzymes may act as endogenous factors which contribute to the increase in VBN. Thus, there is a possibility that VBN detected in the beef samples may not be solely caused by the lactic acid bacteria suggested by metagenomic analysis. Because of these reasons, further research is necessary to clarify the results of this study.
Conclusion
Total aerobic bacteria, Enterobacteriaceae, lactic acid bacteria, Pseudomonas spp., yeast and molds, and psychrotrophic bacteria were primarily detected in beef samples collected in current distribution conditions, and their populations varied among the samples. VBN contents also varied among the beef samples. As samples were categorized with VBNH and VBNL, the metagenomic analysis showed that VBNH samples had a high relative abundance of Lactobacillus and Leuconostoc mostly. Therefore, these results suggest that microorganism populations and VBN varied among beef samples, but specific lactic acid bacteria might be potential bacteria contributing to the production of more VBN in beef. However, the number of samples used was limited and potential VBN production-related bacteria were suggested only by metagenomic analysis, and storage period and storage methods, which may affect the composition of microorganisms, were not examined in this study. Additionally, endogenous enzymes in meat could also contribute to VBN production. Therefore, further research is necessary to identify and isolate these bacteria. This research may involve the inoculation of potential bacteria based on the metagenomic analysis into a larger sample size, followed by the analysis of their VBN production while excluding VBN production by endogenous factors. In further research, an investigation of the bacteria contributing to VBN production under different storage methods and storage periods may also be necessary followed by a comparison of the result with the one in this study. If the design of the research needs to contain additional factors such as temperature, hygiene practices, sanitary conditions in the production process, etc., affecting the compositions of microorganisms in beef that are related to VBN production, the research needs to be conducted with experimental designs of setting levels of these factors with objective standard and with sufficient sample size.