









Introduction
High-caloric intake and low physical activity are the main causes of obesity, which leads to metabolic diseases, such as insulin resistance, type 2 diabetes, and hepatic steatosis (Kahn et al., 2006). Glucose is the primary energy source for the human body, and it primarily provided by food intake. When glucose levels are low in blood, glucose is synthesized by gluconeogenesis to maintain glucose hom- eostasis, since blood glucose is essential for proper brain function (Gerich et al., 2001).
Phosphoenolpyruvate carboxykinase (PEPCK) is a key enzyme in gluconeogenesis that converts oxaloacetate to phosphoenolpyruvate (Stark et al., 2014). In addition, it also plays a key role in glyceroneogenesis, serine synthesis, and the conversion of carbon skeletons in some amino acids. It is known that PEPCK knock-out mice are not able to perform gluconeogenesis in the liver, which leads to hypoglycemia-induced death (Yang et al., 2009). Various hormones and nutritional conditions have been shown to regulate PEPCK expression and activity. Several studies have reported that hepatocyte nuclear factor 4 alpha (HNF4α), glucocorticoid receptor (GR), and Nur77 regulate transcription of PEPCK gene through binding to their cognate response elements located in the PEPCK promoter region (Babeu et al., 2014; Cassuto et al., 2005; Liu et al., 2007; Lu, 2016; Song et al., 2006; Yamamoto et al., 2004). HNF4α (NR2 A1) is a transcription factor belonging to the nuclear receptor superfamily, and is essential for the expression of liver-specific genes (Babeu et al., 2014). HNF4α plays an important role in the regulation of metabolic gene expression in the liver, and also functions as a mediator of hormone, stress, and nutrient signaling (Stoffel et al., 1997). HNF4α participates in glucose metabolism by regulating the expression of genes involved in glucose transport and gluconeogenesis. In particular, HNF4α promotes gluconeogenesis by stimulating PEPCK expression, and its overexpression is known to induce diabetes (Stoffel et al., 1997; Yamagata, 2014).
Recently, many studies have implicated a role for the gut microbiota in energy metabolism and metabolic diseases such as obesity and diabetes (Musso et al., 2010). It has been shown that gut microorganisms regulate host lipid and carbohydrate metabolism, and this function of gut microorganisms is closely associated with the nutritional status of the host (Nagpal et al., 2016). A high-fat diet (HFD) induces a change in the intestinal microflora, which promotes low-level chronic inflammation by increasing lipopolysaccharide (LPS) concentration. Chronic inflammation facilitates metabolic abnormalities in metabolic tissues and eventually leads to metabolic diseases such as obesity and type 2 diabetes (Golozoubova et al., 2001; Keipert et al., 2014).
Probiotics are defined as “live microorganisms that confer health benefits on the host when they are administered in adequate amounts” (Hemarajata et al., 2013). The Lactobacillus acidophilus NS1 (LNS1) used in this study is a probiotic strain that has been shown to improve intestinal and metabolic diseases by restoring a healthy intestinal microbial community. Recently, we observed that LNS1 administration to HFD-fed mice protects against HFD-induced weight gain and increase of fasting glucose level in blood with improved insulin sensitivity.
In this study, we examined the effect of LNS1 on the expression and activity of key transcription factors involved in glucose metabolism to determine how LNS1 regulates glucose metabolism, and we demonstrated that LNS1 inhibits the expression of PEPCK, a key gluconeogenic enzyme, by regulating HNF4α transcriptional activity
Materials and Methods
All animal experimental procedures were conducted in accordance with the protocols approved by approved by Institutional Animal Care and Use Committee of Chonnam National University. Seven-week-old C57BL/6 male mice (weight, 19±2 g) were fed a normal diet (ND; 16% of calories from fat; Damul Science) or a high-fat diet (HFD; 45% of calories from fat; Research Diets, Inc.). Lactobacillus acidophilus NS1 (LNS1) resuspended in PBS (300 μL at ~1.0 × 108 CFU/mL) or vehicle (PBS, 300 μL) was orally administered to mice daily for 12 wk.
HEK293T cells were maintained in Dulbecco’s modified eagle’s medium containing fetal bovine serum (10%) and penicillin/streptomycin (1%). Supernatant was obtained by centrifugation from bacteria culture medium (BCM; de Man, Rogosa, and Sharpe broth) in which LNS1 was incubated for 48 h, and used as LNS1-CM. pGL3-PEPCK promoter-Luciferase and/or HNFα, GR, and Nur77 expression plasmids were transfected into HEK293T cells by using Superfect (Qiagen). After 12 h transfection, BCM or LNS1-CM was treated to the cells at 1:50 dilution and incubated for another 24 h. Luciferase activity was measured with a Luminometer (Berthhold).
Total RNAs were extracted from mouse liver and Hep G2 cells by RiboExTM (GeneAll). cDNA was synthesized using MMLV-RTase (Promega) and Oligo dT primers (Promega). Synthesized cDNA was amplified using eTaq polymerase (Solgent). Gene expression was quantified relative to that of the internal control, 36B4, using a Gel doc XR system (Bio-Rad). Primers for PEPCK and 36B4 have been previously described (16), and the primer sequences are listed in Table 1.
Results
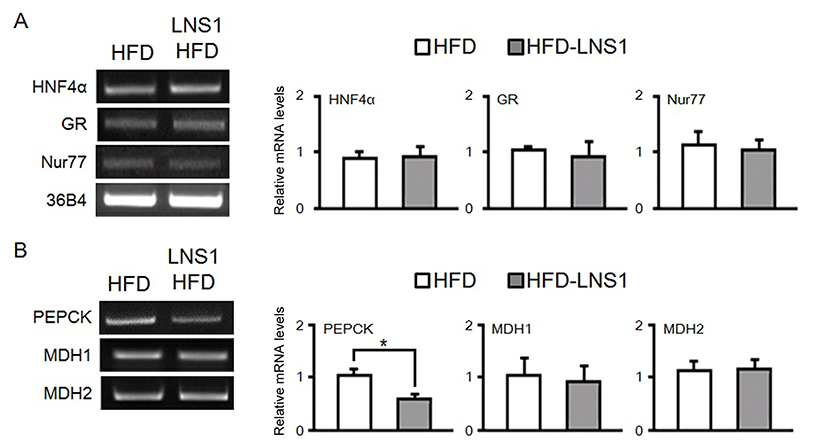
HFD alters glucose homeostasis in part by increasing gluconeogenesis through dysregulated expression of gluconeogenic genes such as PEPCK, which leads to obesity, insulin resistance, and diabetes. We previously observed that administration of LNS1 with HFD to mice reduced HFD-induced body weight gain, increase of blood glucose and improved insulin sensitivity. Since many studies have reported that the expression of transcription factors important for energy metabolism could be changed in metabolic tissues including liver by administration of a HFD, LNS1 may inhibit HFD-induced abnormalities in glucose metabolism by suppressing the altered expression of key transcription factors which regulate transcription of gluconeogenic enzyme genes. To test this hypothesis, we determined the expression of HNF4α, GR, and Nur77, transcription factors important for the regulation of gluconeogenesis, in the livers of HFD-fed mice (HFD mice) and mice fed a HFD with LNS1 (HFD-LNS1 mice). As shown in Fig. 1A, in RT-PCR analysis, administration of LNS1 did not affect mRNA levels of these transcription factor genes in the liver of HFD-fed mice (Fig. 1A). Next, we tested the effect of LNS1 administration on the expression of enzyme genes required for gluconeogenesis. LNS1 dramatically reduced the expression of PEPCK, a key gluconeogenic enzyme, in the liver of HFD mice, while it did not affect the expression of malate dehydrogenase 1 (MD H1) or MDH2 (Fig. 1B).
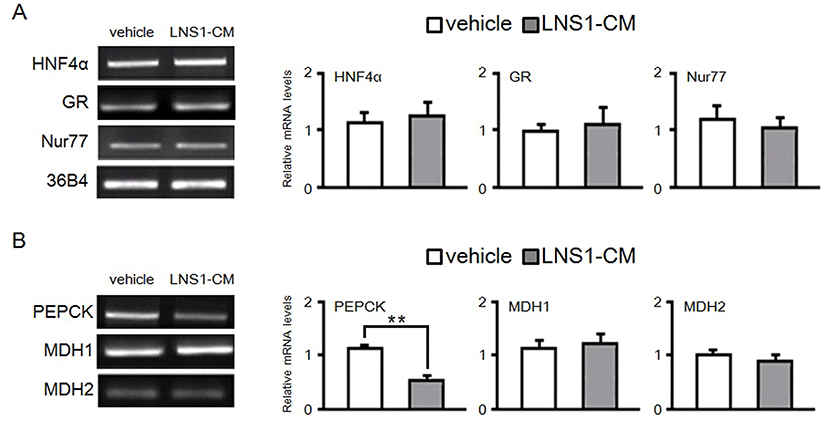
Accumulating evidences suggest that probiotics, including Lactobacillus strains, could change host physiology such as immune response and energy metabolism, and signaling molecules secreted by probiotics could mediate these probiotic effects. We recently observed that treatment of LNS1-CM to HepG2 cells, human hepatoma cells, has similar effect on lipid metabolism compared with LNS1 regulation of hepatic lipid metabolism in mice.
Therefore, to further confirm the regulatory effect of LNS1 on the expression of genes important for gluconeogenesis, we treated LNS1-CM to HepG2 cells for 24 h, and then determined the mRNA levels of these genes using RT-PCR.As shown in Fig. 2A, when HepG2 cells were treated with LNS1-CM, the expression of gluconeogenic transcription factors (HNF4α, GR and Nur77) was not changed. However, LNS1-CM reduced mRNA levels of PEPCK gene in parallel with no change in the expression of MDH1 or MDH2 (Fig. 2B). These results showed that LNS1 specifically regulates hepatic expression of the PEPCK gene, both in vivo and in vitro.
While LNS1 and LNS1-CM reduced the expression of PEPCK, a key gluconeogenic enzyme, in the liver and HepG2 cells, respectively, they did not affect the expression of HNF4α, GR, or Nur77, that are known to promote transcription of PEPCK gene via binding to the PEPCK promoter region. It is possible that LNS1 may regulate transcription of PEPCK by regulating the transcriptional activities of these transcription factors, even though LNS1 did not affect the expression of these transcription factors.
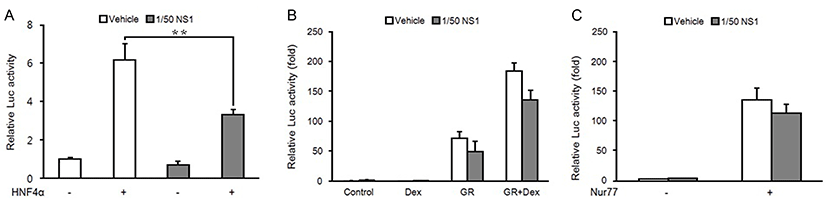
Therefore, we performed a reporter gene assay in HepG2 cells using a luciferase plasmid fused to the mouse PEPCK promoter (pGL3-PEPCK-Luc). As shown in Fig. 3A, when an HNF4α expression plasmid was transfected into HepG2 cells with pGL3-PEPCK-Luc, PEPCK promoter activity was increased about 6-fold compared with the control cells transfected with an empty vector. However, addition of LNS1-CM to HepG2 cells transfected with HNF4α expression plasmid dramatically reduced HNF4α induction of PECPK promoter activity.
In addition, GR, another transcription activator of PEPCK expression, also increased PEPCK promoter activity (Fig. 3B). Furthermore, when dexamethasone, a GR agonist, was added to HepG2 cells transfected with a GR expression plasmid, the transcriptional activity of GR was further increased. Although LNS1-CM treatment mildly reduced GR transcriptional activity in the absence or presence of dexamethasone, its inhibitory effect on GR activity was not significant as compared with its effect on HNF4α activity. Similar to the effect of LNS1-CM on GR transcriptional activity in the reporter assay using pGL3-PEPCK-Luc, LNS1-CM did not significantly inhibit Nur 77-induced PEPCK promoter activity (Fig. 3C). Together, our data demonstrate that LNS1 inhibits the expression of PEPCK gene by specifically inhibiting the transcriptional activity of HNF4α.
Discussion
Modern lifestyle, characterized by high caloric intake with low physical activity, is closely associated with abnormality in glucose homeostasis, which leads to metabolic diseases, such as obesity, insulin resistance, and diabetes (Kahn et al., 2006).
Accumulating evidence suggests that high-fat, high-carbohydrate diets rapidly change the intestinal microflora, which are important for metabolic homeostasis, including glucose metabolism (Rosa et al., 2012; You et al., 2015). Probiotics have been known to be important for intestinal microbial balance and improve inflammatory bowel diseases, including infectious diarrhea and irritable bowel syndrome (Kvasnovsky et al., 2015; Mazurak et al., 2015). In addition, probiotics have shown some beneficial effects on various metabolic diseases. Many studies have reported that administration of probiotics effectively reduces HFD-induced obesity, insulin resistance, and hepatic steatosis (You et al., 2015).
We observed that oral administration of LNS1 to mice fed a HFD reduces HFD-induced gains of body weight and liver and adipose tissue weights. In addition, LNS1 inhibited increase of blood glucose levels due to HFD feeding, with improvement of insulin sensitivity. In this study, to identify the molecular mechanism of LNS1 regulation of glucose metabolism in HFD-fed mice, we examined LNS1 effect on the expression of gluconeogenic genes and transcription factors that regulate the expression of genes involved in gluconeogenesis. Although LNS1 and LNS1-CM did not affect the expression of two gluconeogenic genes (MDH1 and MDH2), and key transcription factors (HNF4α, GR and Nur77) for glucose homeostasis in the liver and HepG2 cells, LNS1 and LNS1-CM significantly reduced HFD induction of PEPCK expression in the liver and HepG2 cells, respectively. Furthermore, in the reporter gene assay using a reporter plasmid fused to the PEPCK promoter region, LNS1-CM suppressed HNF4α transcriptional activity without any significant effect on GR and Nur77 transcriptional activities, suggesting that LNS1 may improve HFD-induced alteration of glucose metabolism, at least in part, through inhibition of the expression of PEPCK gene, a key enzyme for gluconeogenesis, by regulating the transcriptional activity of HNF 4α.
At present, we do not know how LNS1 inhibits the transcriptional activity of HNF4α. Gut microorganisms, including Lactobacillus strains, have been shown to produce short-chain fatty acids, such as acetate, propionate, and butyrate, from dietary fiber and low-digestible polysaccharides through fermentation (Jansen et al., 2004). Interestingly, recent studies reported that some of these short-chain fatty acids stimulate various signaling pathways important for energy homeostasis. Therefore, it is possible that LNS1 may regulate the transcriptional activity of HNF 4α by producing these molecules. Further study will be required to clearly define the molecular mechanism underlying LNS1 regulation of HNF4α activity. We are currently searching for the signaling molecules produced by LNS1 that regulate metabolic signaling pathways, including HNF4α.
Our study, together with our previous study, demonstrates that administration of L. acidophilus NS1 may be an effective preventive strategy for controlling insulin resistance and type 2 diabetes, which are closely associated with altered glucose metabolism.